Clear Sky Science · es
haCCA: integración multimódulo de transcriptomas y metabolomas espaciales basados en puntos
Por qué importa mapear las moléculas in situ
Nuestros cuerpos están formados por innumerables pequeños vecindarios de células, cada uno con su propia combinación de genes activos y sustancias químicas. Hasta hace poco, los científicos tenían que estudiar estas moléculas tras triturar el tejido en una pasta homogénea, perdiendo por completo la noción de “dónde” se encontraban. Este artículo presenta un nuevo método computacional, llamado haCCA, que une dos potentes técnicas de imagen para que los investigadores puedan ver, in situ, cómo se distribuyen los genes y las moléculas pequeñas a lo largo de tejidos y tumores reales. Ese tipo de mapa puede revelar patrones ocultos de enfermedad y sugerir tratamientos más precisos.
Dos vistas diferentes del mismo tejido
El estudio se centra en combinar datos de dos métodos espaciales cada vez más usados en biología. La transcriptómica espacial registra qué genes están activados en miles de pequeños puntos a lo largo de una lámina de tejido. La imagen por espectrometría de masas MALDI cuantifica muchas moléculas pequeñas, como metabolitos y lípidos, en paños de puntos con densidades similares. El problema es que estos dos instrumentos no miden exactamente las mismas posiciones ni el mismo conjunto de características, por lo que sus datos son como dos mapas desalineados con leyendas diferentes. Los enfoques existentes suelen intentar emparejar las formas de las secciones tisulares basándose únicamente en sus coordenadas, lo que puede ser impreciso y carecer de un modo de comprobar realmente qué tan bien funcionó la alineación.
Una forma más inteligente de alinear mapas moleculares
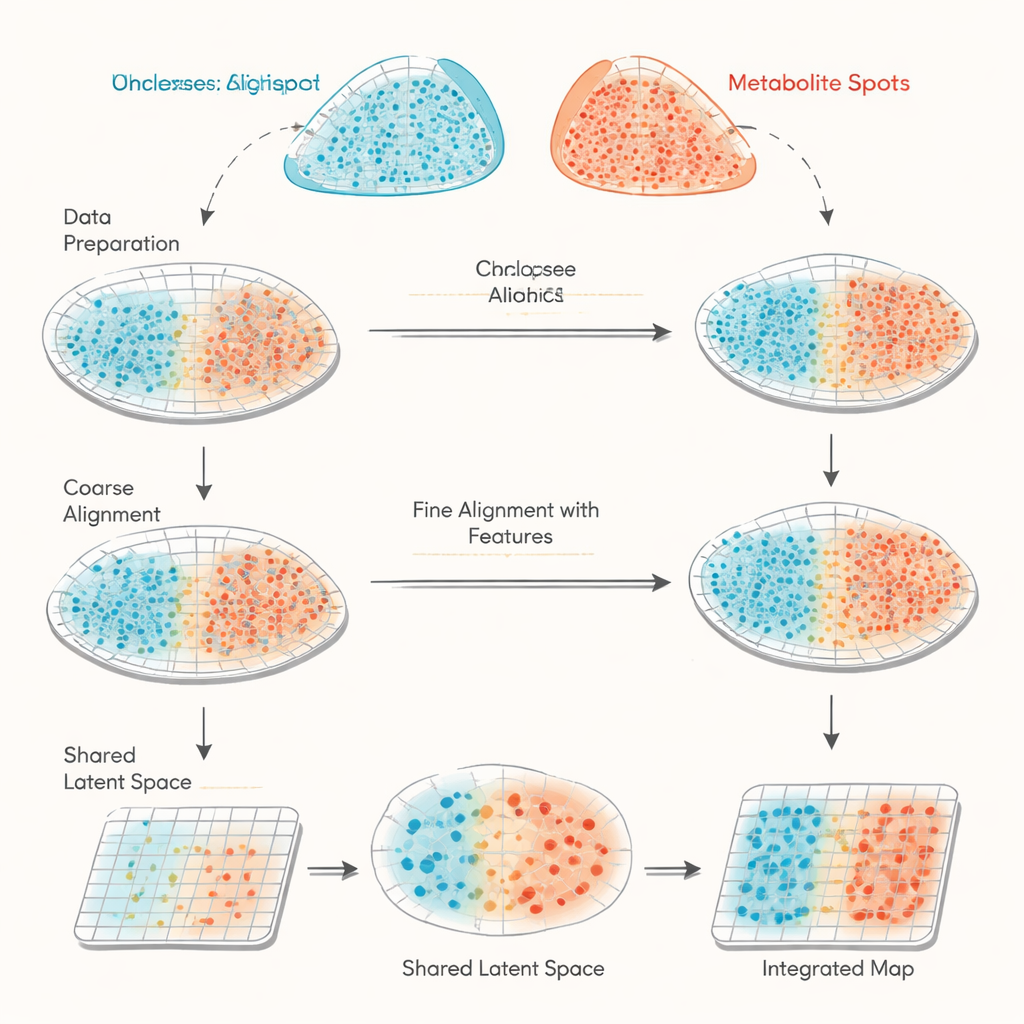
haCCA (acrónimo de análisis canónico correlacional guiado por anclas y jerárquico) aborda este desafío combinando geometría y biología. Primero realiza una “alineación morfológica” en dos pasos de las rejillas de puntos de las dos tecnologías. Expertos humanos señalan algunos hitos coincidentes en las imágenes tisulares para corregir a grandes rasgos desplazamientos y rotaciones, y luego un paso automatizado ajusta los valores atípicos cerca de bordes rasgados o piezas ausentes. A continuación, el método busca pares de “anclas”: puntos cercanos en el espacio y situados en regiones localmente uniformes, lo que los hace probables representantes de la misma área tisular. A partir de estas anclas, haCCA calcula qué genes y metabolitos tienden a variar conjuntamente y los reduce a una representación compartida de baja dimensión que captura sus patrones conjuntos más fuertes.
Transformar correlaciones en una imagen tisular unificada
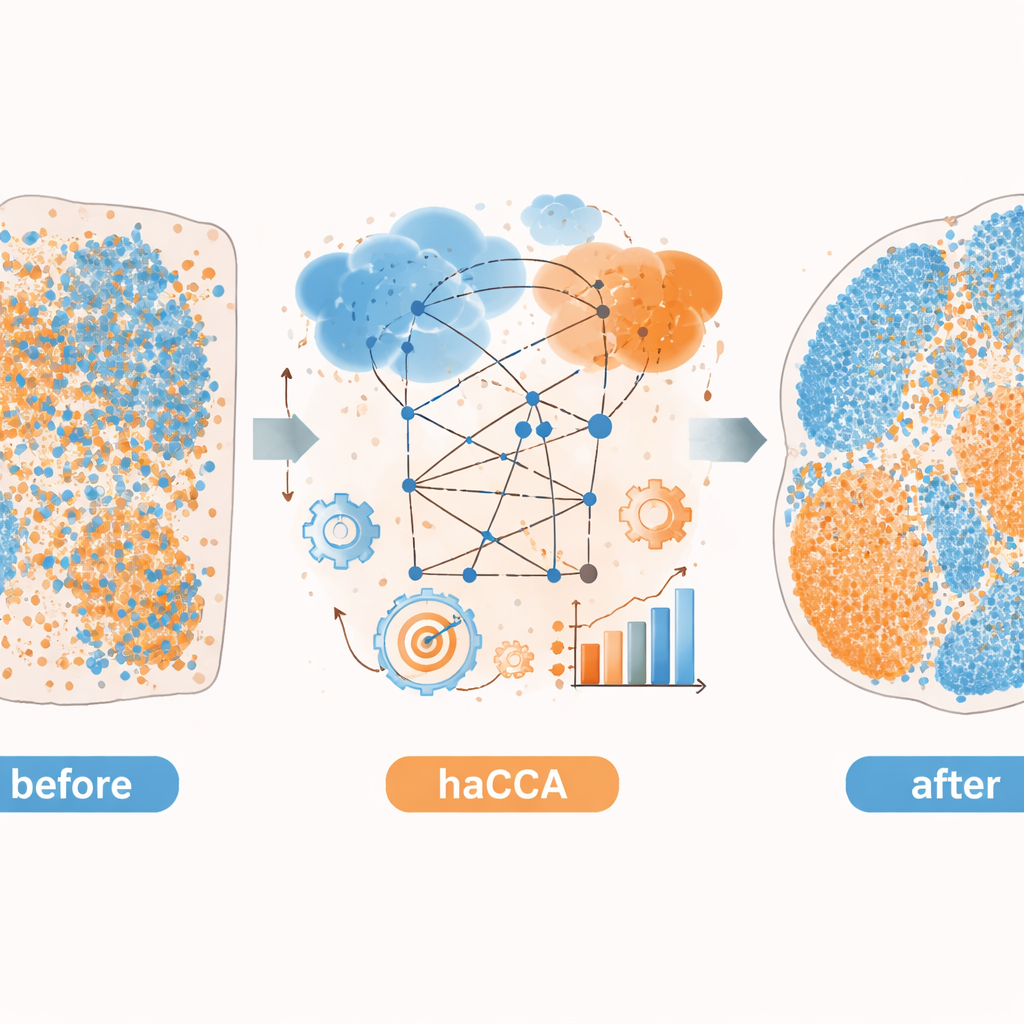
Con las coordenadas espaciales y la representación molecular compartida, haCCA resuelve un problema de optimización para decidir cuán probable es que cada punto génico se corresponda con cada punto de metabolitos. Este paso está diseñado para mantener los puntos próximos en el espacio pero también similares en su perfil combinado gen‑metabolito. El resultado final es un “plan de transporte” que vincula cada punto de un conjunto de datos con su mejor pareja en el otro, produciendo un mapa integrado multimodal. En datos de prueba cuidadosamente construidos —donde se conocen las relaciones verdaderas— los autores muestran que cada etapa del flujo de trabajo (alineación gruesa, alineación refinada y emparejamiento sensible a características) mejora de forma constante tres medidas independientes de precisión. En comparación con otras herramientas que se basan principalmente en la geometría, haCCA logran de manera consistente una alineación superior y una transferencia más fiel de las etiquetas regionales.
Revelando biología oculta en cáncer cerebral y hepático
Los autores aplican luego haCCA a tejidos reales de cerebro de ratón y tumores hepáticos. Para el cerebro, integran datos comerciales de transcriptómica espacial con imágenes de metabolitos procedentes de la misma sección o de secciones vecinas. El método preserva territorios metabólicos conocidos y reconstruye solapamientos esperados, como la colocalización de la dopamina con el gen que codifica su enzima clave. Mediante la agrupación conjunta de genes y metabolitos, encuentran que los datos combinados separan subregiones tisulares más matizadas que cualquiera de las modalidades por sí sola. En un modelo preclínico de colangiocarcinoma intrahepático —un tipo de cáncer de hígado— usan haCCA para comparar tumores que pueden o no formar trampas extracelulares de neutrófilos—estructuras en forma de red liberadas por células inmunitarias. Los mapas integrados revelan que, cuando estas trampas están presentes, un gen llamado Scd1 y sus ácidos grasos asociados se enriquecen en regiones malignas, lo que apunta a un cambio hacia un metabolismo lipídico alterado en el tumor.
Qué significa esto para la investigación futura
En términos cotidianos, haCCA es como alinear fotos aéreas tomadas con cámaras diferentes—una sensible a los contornos de los edificios, la otra a las firmas térmicas—para obtener una imagen más nítida de lo que ocurre en cada manzana. Al fusionar con precisión dónde están activos los genes con dónde se acumulan metabolitos clave, este flujo de trabajo ayuda a los científicos a perfilar a la vez ambos lados del comportamiento celular: las instrucciones y la química resultante. El enfoque mejora los métodos de alineación previos, está empaquetado en una herramienta accesible en Python y puede extenderse a otras tecnologías espaciales. A medida que estos mapas integrados se vuelvan más habituales, podrían profundizar nuestra comprensión de cómo los tumores y otros tejidos organizan su metabolismo, responden al tratamiento y evolucionan con el tiempo.
Cita: Xu, J., Shen, XT., Zhang, C. et al. haCCA: multi-module Integration of spot-based spatial transcriptomes and metabolomes. Commun Biol 9, 248 (2026). https://doi.org/10.1038/s42003-026-09526-w
Palabras clave: multi-ómicas espaciales, transcriptómica, metabolómica, metabolismo tumoral, integración de datos