Clear Sky Science · es
Determinación ab initio de las estabilidades de fase de sólidos con desorden dinámico: desorden rotacional C2 en Li2C2
Por qué importa este sólido cambiante
Muchas tecnologías modernas dependen de sólidos que pueden cambiar discretamente su estructura interna al calentarse o comprimirse. Estos cambios, denominados transiciones de fase, son clave en ideas como la refrigeración en estado sólido y baterías más seguras. Este estudio analiza un compuesto simple, el carburo de litio (Li2C2), que pasa de una forma ordenada a otra más inquieta y con desorden dinámico al aumentar la temperatura. Al observar esta transformación átomo por átomo en simulaciones por ordenador, los autores muestran cómo la «inquietud» interna de pequeñas unidades moleculares puede inclinar la balanza entre dos estructuras cristalinas.
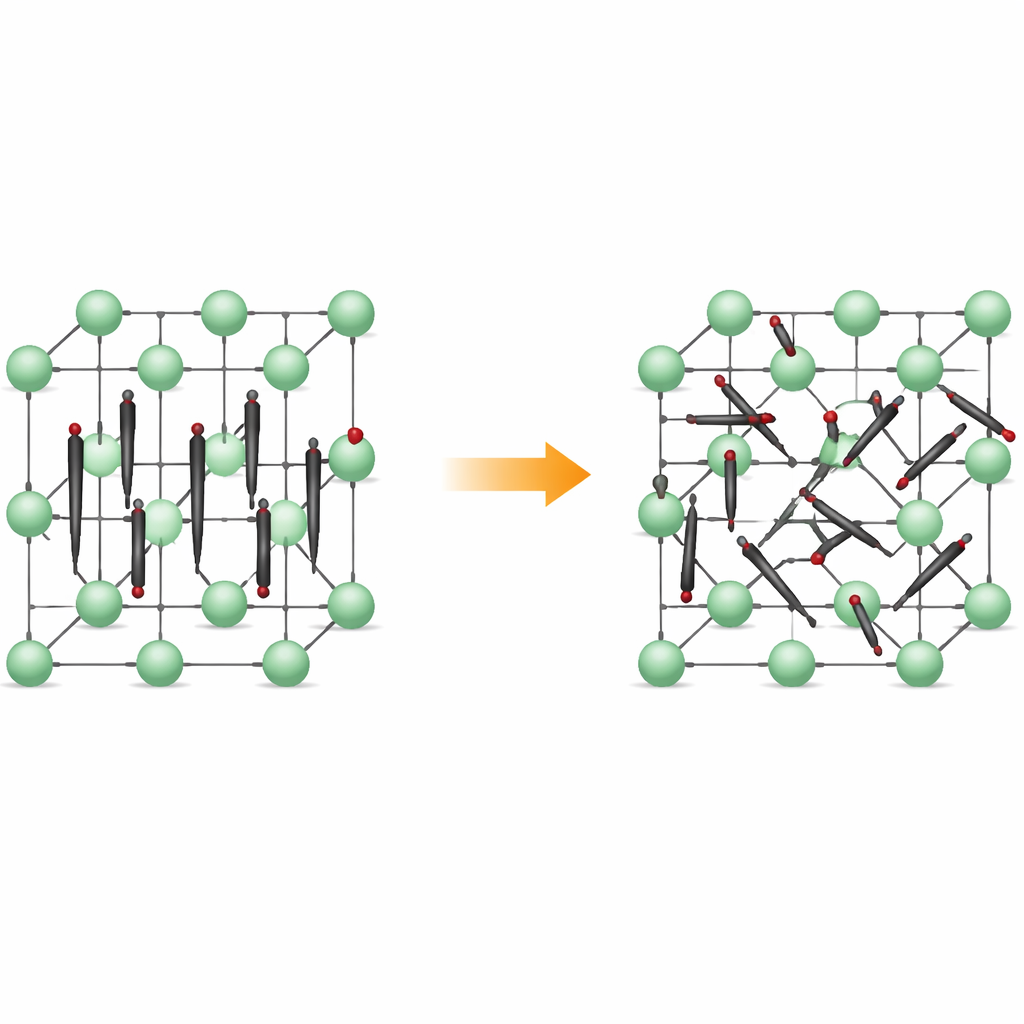
De filas ordenadas a movimiento inquieto
A bajas temperaturas, Li2C2 forma un cristal ortorrómbico: sus átomos de carbono se emparejan en pequeños dímeros C2 que apuntan casi todos en la misma dirección, como cerillas alineadas. Los iones de litio se sitúan entre ellos, creando una trama tridimensional regular. Al calentar el material, se transforma en una forma cúbica, donde las posiciones de los centros de los dímeros permanecen ordenadas en una red, pero los propios dímeros ya no mantienen una dirección fija. En lugar de ello, rotan entre varias orientaciones preferidas, permaneciendo en valles de energía poco profundos correspondientes a alineamientos concretos. El material sigue siendo sólido, pero su estructura interna adquiere desorden dinámico.
Siguiendo el cambio a lo largo de un camino suave
Para entender qué fase es más estable a una temperatura dada, hay que comparar sus energías libres, que combinan energía y entropía (una medida del desorden). Los métodos estándar basados en pequeñas vibraciones alrededor de posiciones fijas tienen dificultades cuando los átomos se desplazan o rotan de forma significativa. Aquí, los autores usan una técnica llamada integración termodinámica esfuerzo–deformación, basada en dinámica molecular de primeros principios. Construyen un camino de deformación suave que transforma continuamente la celda de simulación desde la estructura ortorrómbica de baja temperatura hasta la cúbica de alta temperatura. A lo largo de ese camino, ejecutan simulaciones largas a temperatura fija y miden cómo responde el esfuerzo interno a la deformación impuesta. Integrar esta respuesta de esfuerzo proporciona la diferencia de energía libre entre las dos fases.
Ver la entropía a través del movimiento atómico
Los cálculos revelan que alrededor de 600 K la fase ortorrómbica de baja temperatura aún está ligeramente favorecida, mientras que a 650 K la fase cúbica gana por unos pocos milésimos de electrón-voltio por unidad formulada. Interpolar entre estos resultados da una temperatura de transición de aproximadamente 611 K. Esto es más bajo que las estimaciones experimentales, pero sigue en razonable acuerdo, dada la pequeña magnitud de las diferencias de energía libre implicadas. La energía interna de la fase cúbica es en realidad mayor; lo que la estabiliza es una gran ganancia de entropía, trazada directamente al desorden rotacional de los dímeros C2. Al analizar cómo la orientación de cada dímero pierde memoria de su dirección inicial con el tiempo, los autores muestran que los dímeros se reorientan en escalas de tiempo sub-picosegundo, difuminando la línea entre las categorías usuales de entropía «vibracional» y «configuracional».
Más allá de imágenes simples del desorden sólido
El trabajo también pone de manifiesto que atajos comunes —como tratar la entropía como una suma simple de vibraciones alrededor de configuraciones fijas más un conteo separado de orientaciones estáticas— se rompen para materiales como Li2C2. Dado que las rotaciones de los dímeros son rápidas y están fuertemente acopladas a las vibraciones ordinarias, el sistema no puede dividirse nítidamente en partes «vibrantes» y «reordenantes». El método de integración esfuerzo–deformación sortea esta dificultad: extrae la energía libre completa directamente de la dinámica microscópica, sin necesidad de suponer cómo debe repartirse la entropía.
Lo que nos enseña el estudio
En términos cotidianos, el estudio muestra cómo un sólido puede permanecer rígido mientras sus bloques internos de construcción ganan libertad para girar y moverse, y cómo esa libertad interna puede hacer que una estructura más desordenada sea preferida termodinámicamente. En Li2C2, la fase cúbica de alta temperatura se estabiliza no porque sea energéticamente más barata, sino porque ofrece muchas más maneras para que los dímeros C2 se orienten y se muevan. Al demostrar que la integración termodinámica esfuerzo–deformación puede captar este sutil equilibrio entre orden, energía y entropía, el trabajo abre un camino para predecir transiciones similares en otros sólidos con desorden dinámico que podrían sustentar futuros dispositivos de refrigeración, baterías y materiales inteligentes.
Cita: Klarbring, J., Filippov, S., Häussermann, U. et al. Ab initio determination of phase stabilities of dynamically disordered solids: rotational C2 disorder in Li2C2. Sci Rep 16, 8965 (2026). https://doi.org/10.1038/s41598-026-43795-z
Palabras clave: transición de fase en estado sólido, desorden dinámico, dinámica molecular, carburos de litio, integración termodinámica