Clear Sky Science · es
Modelo profundo híbrido afinado para el diagnóstico del cáncer de mama usando datos genéticos
Por qué esto importa para pacientes y familias
El cáncer de mama es hoy el tumor diagnosticado con más frecuencia en mujeres en todo el mundo, y detectarlo pronto puede marcar la diferencia entre la vida y la muerte. Los médicos disponen cada vez más de la información genética de las personas, pero convertir decenas de miles de mediciones génicas en respuestas claras es extraordinariamente difícil. Este artículo describe un nuevo modelo computacional que interpreta estos patrones genéticos complejos para detectar el cáncer de mama y predecir resultados con una precisión notable, ofreciendo potencialmente a los clínicos un asistente potente para tomar decisiones más tempranas y fiables.
De los genes a las señales de alerta
Cada tumor mamario porta una huella molecular codificada en la actividad de miles de genes. Los autores se propusieron construir un sistema capaz de leer directamente esa huella, en lugar de basarse solo en imágenes o en un puñado de genes bien conocidos como BRCA1 y BRCA2. Trabajaron con dos de los mayores recursos públicos en genómica del cáncer: la cohorte de cáncer de mama de TCGA, que incluye la actividad de 17.814 genes en 590 muestras, y el estudio METABRIC, que contiene información genómica y clínica de más de 1.400 pacientes. Su objetivo fue ambicioso: diseñar un método que pudiera manejar este aluvión de información, encontrar las señales más reveladoras y funcionar con fiabilidad en grupos de pacientes completamente independientes.
Reducir miles de genes a un conjunto útil
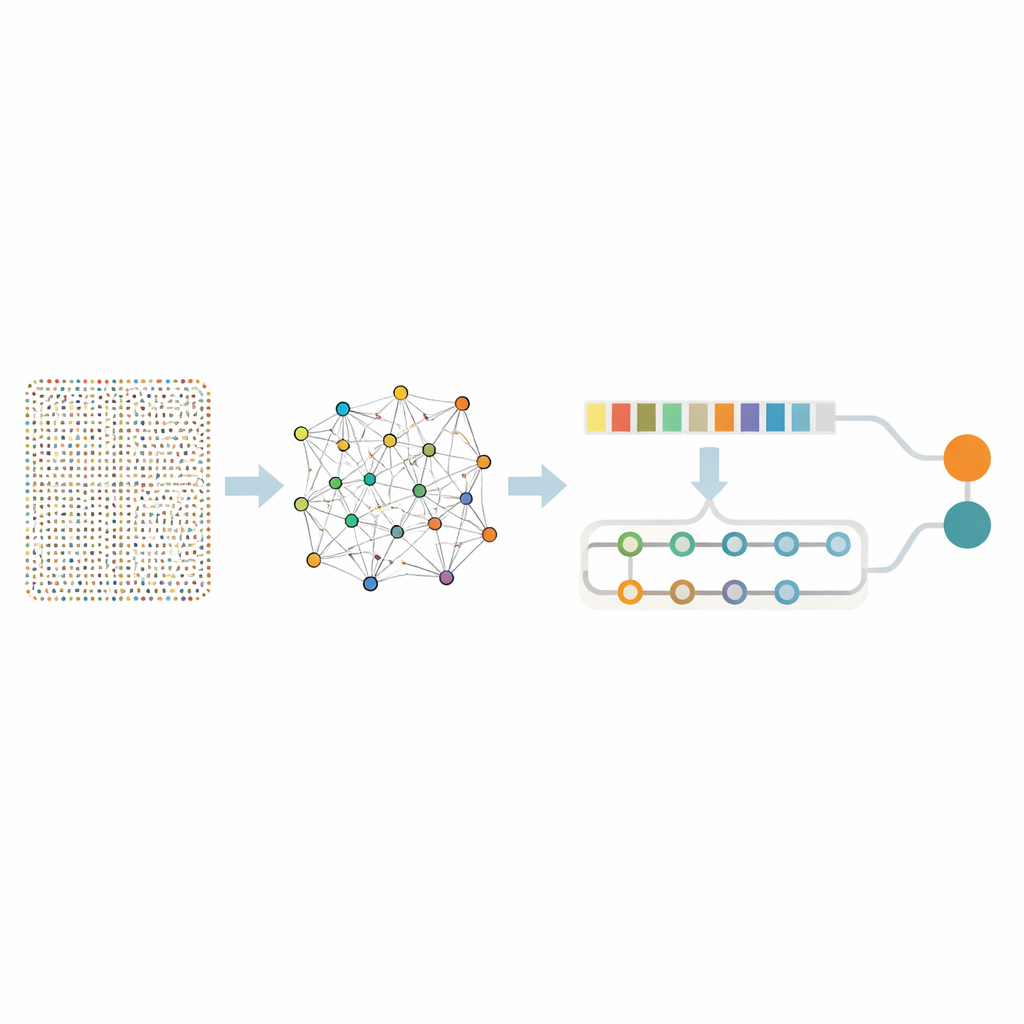
Analizar casi dieciocho mil genes de una vez es abrumador incluso para algoritmos avanzados, y existe el riesgo de captar ruido sin significado. Por ello, los investigadores emplearon un “tamiz” en dos pasos para aislar un conjunto más pequeño de genes realmente informativos. Primero aplicaron una técnica llamada Random Forest, que plantea muchas preguntas a varios árboles de decisión sobre qué genes son más relevantes para distinguir tejido canceroso de muestras sanas. Ese paso redujo la lista a 436 genes prometedores. A continuación examinaron cómo se comportan esos genes en conjunto mediante minería de reglas de asociación, un método que detecta grupos de genes que tienden a activarse al mismo tiempo en los tumores. Esta capa adicional de análisis identificó pares y redes génicas vinculados a procesos clave del cáncer, como la división celular rápida, la reparación del daño en el ADN y cambios en el tejido circundante al tumor. Tras esta depuración, quedaron 332 genes: todavía ricos en significado biológico, pero mucho más manejables para un análisis más profundo.
Una red neuronal en dos partes que aprende patrones y contexto
Con este conjunto focalizado de genes, el equipo construyó un modelo híbrido de aprendizaje profundo que combina dos tipos de redes neuronales. Una parte, conocida como red convolucional, recorre la lista de genes para captar patrones locales: racimos de genes que tienden a subir o bajar juntos. La segunda parte, una red de memoria bidireccional, analiza la misma información manteniendo el seguimiento de relaciones a largo alcance, capturando cómo genes distantes se influyen entre sí a lo largo del perfil completo. Antes del entrenamiento, los autores equilibraron los datos para que las muestras cancerosas y no cancerosas estuvieran representadas de forma justa y añadieron pequeñas cantidades de ruido artificial, enseñando al modelo a no dejarse engañar por fluctuaciones aleatorias.
Rendimiento del sistema en pruebas del mundo real
Cuando se entrenó y evaluó con datos de TCGA, la red híbrida distinguió correctamente tumores de muestras normales con aproximadamente un 97 % de precisión y una capacidad casi perfecta para separar ambos grupos. Es importante destacar que superó a configuraciones más sencillas de aprendizaje profundo y a herramientas estándar de aprendizaje automático como la regresión logística y las máquinas de vectores de soporte, incluso cuando esos métodos competidores recibieron los mismos genes cuidadosamente seleccionados. La prueba más exigente, sin embargo, fue si el modelo resistiría en un conjunto de datos totalmente distinto. Aplicado a METABRIC, recogido en otros hospitales y con métodos de laboratorio diferentes, el sistema mantuvo un alto rendimiento: en su mejor ejecución alcanzó un 99,3 % de precisión e identificó correctamente a todos los pacientes que más tarde fallecieron por cáncer de mama, una propiedad crucial si la herramienta se va a usar para señalar casos de alto riesgo.
Qué podría significar esto para la atención futura
Para un público no especialista, la conclusión es que este estudio ofrece un filtro y lector inteligente para datos genéticos capaz de detectar cáncer de mama y riesgos asociados con una consistencia notable en grandes grupos de pacientes. Al combinar una estrategia reflexiva de selección génica con una red neuronal de dos ramas, los autores demuestran que los ordenadores pueden extraer señales clínicamente relevantes de conjuntos genéticos enormes, no solo en un estudio sino a través de cohortes independientes. Aunque se necesita más trabajo para probar el enfoque en poblaciones diversas y para explicar sus decisiones en detalle, el método apunta hacia un futuro en el que una simple muestra de sangre o tejido podría alimentarse en modelos así y ayudar a los médicos a detectar tumores antes y a ajustar el tratamiento con mayor precisión.
Cita: Hesham, F., Abbassy, M.M. & Abdalla, M. Hybrid tuned deep learning model for breast cancer diagnosis using genetic data. Sci Rep 16, 9664 (2026). https://doi.org/10.1038/s41598-026-41643-8
Palabras clave: genómica del cáncer de mama, diagnóstico mediante aprendizaje profundo, biomarcadores de expresión génica, detección temprana del cáncer, apoyo a la decisión clínica