Clear Sky Science · es
Descubrimiento de hidroxi-triazol como posible inhibidor de la glicoxalasa‑I mediante técnicas de diseño asistido por ordenador
Por qué detener a un pequeño limpiador celular podría combatir el cáncer
Las células cancerosas con frecuencia crecen tan deprisa que se ahogan en sus propios desechos. Uno de sus trucos de supervivencia es un sistema de limpieza integrado que desintoxica subproductos nocivos de la combustión de azúcares. Este estudio explora cómo apagar a un miembro clave de ese sistema, una enzima llamada glicoxalasa‑I, usando ordenadores para cribar decenas de miles de moléculas y experimentos para probar los mejores candidatos. El objetivo es descubrir nuevos “puntos de partida” para fármacos que algún día podrían ayudar a los médicos a envenenar selectivamente las células cancerosas desde dentro.
Un sistema oculto de gestión de desechos dentro de nuestras células
Cada célula descompone constantemente azúcar para producir energía, y este proceso genera un químico reactivo de desecho llamado metilglioxal. En cantidades normales, nuestro cuerpo convierte el metilglioxal en ácido láctico inofensivo mediante el sistema de glicoxalasa, una vía de dos pasos que depende del cofactor glutatión. La glicoxalasa‑I es el primer y más crucial escalón de esta cadena. Las células cancerosas, que consumen azúcar a un ritmo frenético, dependen en gran medida de la glicoxalasa‑I para evitar que el metilglioxal alcance niveles tóxicos. Si se bloquea esta enzima, el metilglioxal se acumula y puede empujar a las células dañadas hacia la muerte programada. Eso convierte a la glicoxalasa‑I en un objetivo atractivo para fármacos anticancerígenos que atacan una vulnerabilidad básica del metabolismo tumoral.
Buscando en el espacio químico con silicio y estadística
En lugar de probar sustancias al azar en el laboratorio, los investigadores utilizaron diseño de fármacos asistido por ordenador para rastrear una gran colección comercial de más de 50.000 pequeñas moléculas. Software especializado limpió y estandarizó cada molécula, luego predijo su forma 3D y comportamiento a un pH similar al corporal. Una etapa rápida de cribado virtual puntuó qué tan bien podría encajar cada candidato en el sitio activo de la glicoxalasa‑I. El equipo aplicó después reglas sencillas sobre tamaño, solubilidad y otras propiedades de afinidad farmacológica para descartar moléculas poco probables de funcionar en el organismo. Un programa de docking más detallado examinó cómo las moléculas más prometedoras podrían orientarse dentro de la enzima, en especial cómo podrían alcanzar y sujetar el átomo de zinc que ocupa el centro de la química de la glicoxalasa‑I.
Una nueva forma de agarrar el núcleo metálico de la enzima
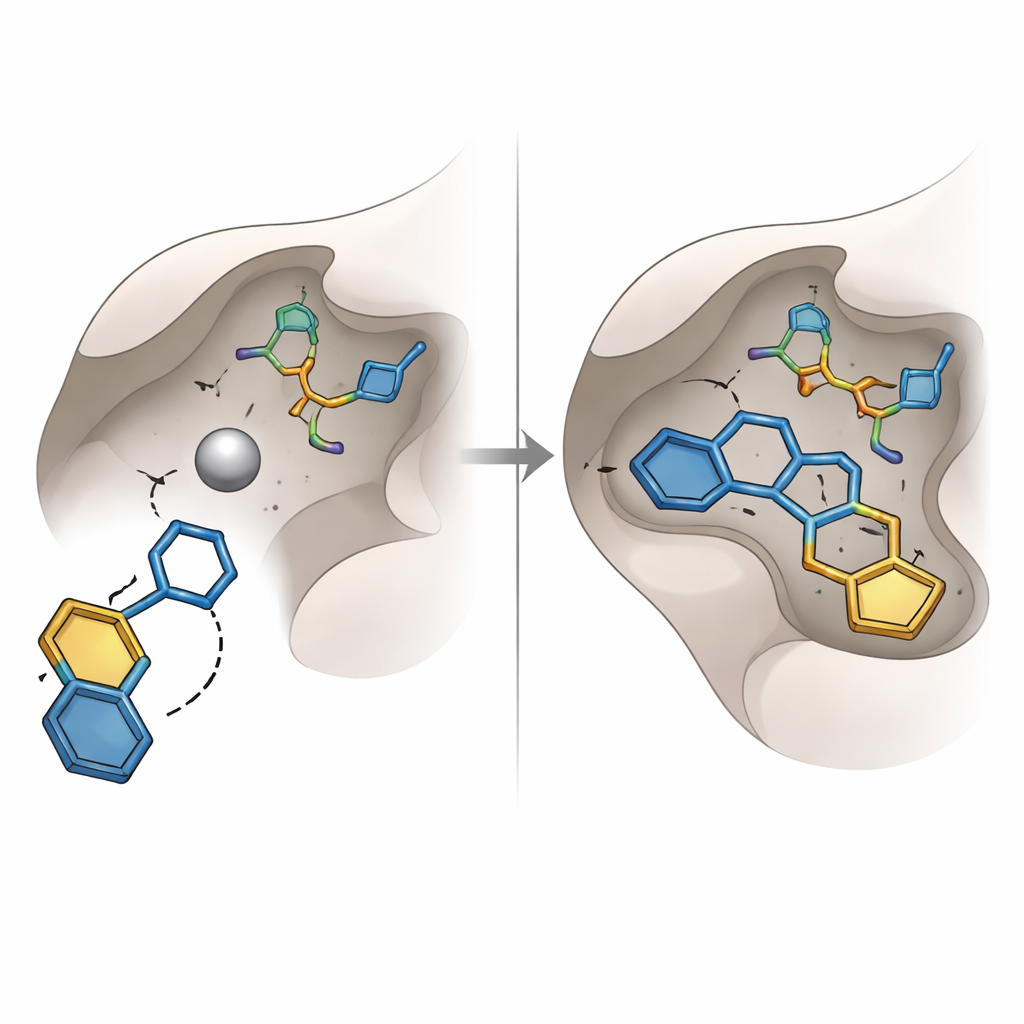
Esfuerzos anteriores para bloquear la glicoxalasa‑I se centraron en grupos químicos conocidos, como ácidos carboxílicos e hidroxámicos, que se unen bien a metales pero a menudo presentan mala estabilidad o efectos secundarios indeseables. El presente estudio, en cambio, descubrió otro tipo de unidad “agarrametales”: un anillo hidroxi‑triazol. Entre dieciséis moléculas mejor clasificadas elegidas para compra y pruebas en laboratorio, una que porta este anillo—codificada SPB07393SC—surgió como la más destacada. En el docking virtual, su grupo hidroxi‑triazol alcanzó el átomo de zinc, mientras que sus dos anillos aromáticos se alojaron en bolsillos oleosos cercanos de la enzima. Simulaciones por ordenador del complejo durante decenas de nanosegundos sugirieron que la molécula permanecía firmemente unida, con distancias estables, una conformación compacta de la proteína y una red persistente de enlaces de hidrógeno.
Poner a prueba las predicciones
Para comprobar si los modelos computacionales se traducían en efectos reales, el equipo midió cuánto ralentizaban las moléculas seleccionadas la actividad de la glicoxalasa‑I humana purificada en un ensayo en placa. Quince de las dieciséis candidatas mostraron solo inhibición débil o insignificante en las condiciones probadas, lo que pone de relieve las trampas de fiarse únicamente de las puntuaciones de docking estático. En contraste, SPB07393SC inhibió la enzima con fuerza, con una potencia en el rango medio de micromolar que la convierte en un “hit” inicial sólido más que en un fármaco acabado. Herramientas informáticas adicionales predijeron que esta molécula debería tener solubilidad aceptable, buena absorción, capacidad para alcanzar el cerebro si fuera necesario y baja probabilidad de provocar ciertas toxicidades genéticas o hepáticas, aunque estas predicciones de seguridad requieren todavía confirmación experimental.
Qué significa esto para futuros fármacos contra el cáncer
El trabajo introduce al hidroxi‑triazol como una nueva forma de anclar candidatos farmacológicos al átomo de zinc en el núcleo de la glicoxalasa‑I, ampliando el repertorio de recursos químicos disponibles para los diseñadores de fármacos. Si bien la SPB07393SC en sí misma es solo un punto de partida, su combinación de capacidad para bloquear la enzima, comportamiento farmacológico predicho y unión estable en simulaciones dinámicas la señalan como un andamiaje prometedor para un mayor ajuste. Más en general, el estudio muestra tanto las fortalezas como los límites del cribado guiado por ordenador: puede reducir rápidamente vastas bibliotecas químicas a unos pocos contendientes realistas, pero los experimentos de laboratorio cuidadosos siguen siendo esenciales para revelar qué moléculas desactivan verdaderamente la enzima de la que dependen las células cancerosas para gestionar sus desechos tóxicos.
Cita: Al-Qazzan, M., Al-Balas, Q., Alnajjar, B. et al. Discovery of hydroxytriazole as a potential glyoxalase-I inhibitor utilizing computer-aided drug design techniques. Sci Rep 16, 9945 (2026). https://doi.org/10.1038/s41598-026-40497-4
Palabras clave: glicoxalasa I, metabolismo del cáncer, diseño de fármacos asistido por ordenador, inhibidores con afinidad al zinc, docking molecular