Clear Sky Science · es
DPAS: puntuación de anomalía de péptidos asociados a la enfermedad para identificar péptidos patógenos mediante aprendizaje de una sola clase
Por qué importan las piezas diminutas de las proteínas para nuestra salud
Los péptidos—tramos cortos de proteínas—se han convertido en protagonistas emergentes de la medicina moderna. Pueden actuar como mensajeros precisos en el organismo y se usan cada vez más como fármacos y marcadores de enfermedad. Sin embargo, determinar qué péptidos están realmente vinculados a la enfermedad suele depender de contar con ejemplos claros tanto de péptidos “enfermos” como de “no enfermos”, algo que la biología rara vez ofrece. Este estudio presenta una nueva manera de detectar péptidos potencialmente dañinos usando únicamente aquellos que ya sabemos que están implicados en la enfermedad, lo que ofrece una vía más rápida y menos sesgada para descubrir futuros diagnósticos y tratamientos.
El reto de encontrar el grupo de “no enfermos”
Los modelos informáticos tradicionales aprenden comparando dos bandos: ejemplos positivos conocidos por su relación con la enfermedad y ejemplos negativos que se consideran inofensivos. En la investigación de péptidos, este segundo grupo es problemático. Muchos péptidos simplemente no han sido probados, por lo que etiquetarlos como “no enfermos” puede inducir a error e introducir sesgos. Estudios previos sobre péptidos anticancerígenos o antiinflamatorios alcanzaron precisiones impresionantes, pero a menudo dependieron de conjuntos de datos negativos construidos a mano o supuestos. Como resultado, sus modelos pueden tener dificultades con señales raras o con nuevos tipos de péptidos asociados a enfermedad que no se parecen a los datos de entrenamiento.
Aprender de lo que conocemos, en lugar de de lo que suponemos
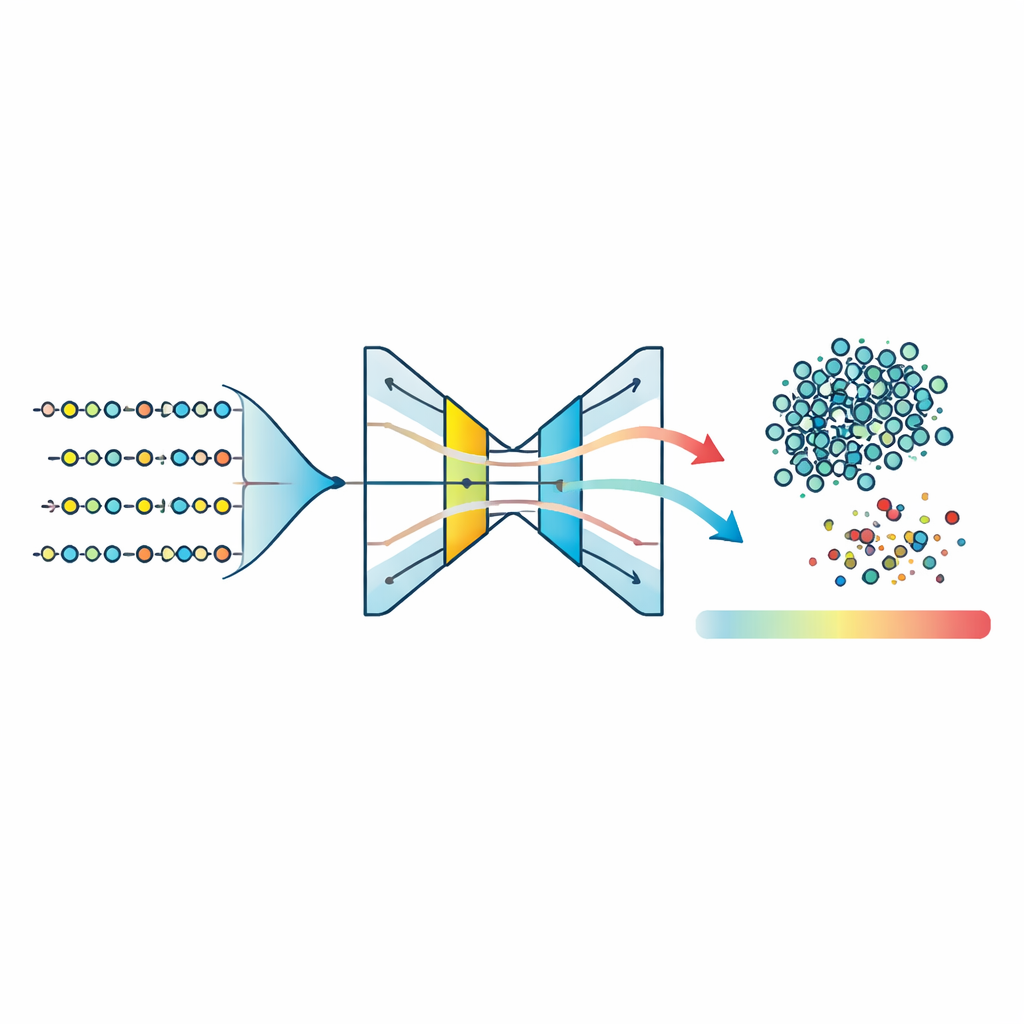
Los autores siguen un camino distinto: en lugar de forzar un problema de dos lados, tratan a los péptidos asociados a la enfermedad como un grupo coherente y se preguntan: «¿Cómo se ve este grupo en detalle?» Reúnen más de 760.000 péptidos humanos mutados de una base de datos especializada en cáncer y describen cada péptido usando un conjunto amplio de características. Estas incluyen la frecuencia de cada aminoácido, la disposición de pares de aminoácidos, rasgos físico‑químicos básicos como el volumen y la hidrofilicidad, y patrones de secuencia cortos recurrentes conocidos como motivos. Una técnica llamada análisis de componentes principales comprime esta descripción de alta dimensión a una forma más manejable conservando las principales fuentes de variación.
Detectar péptidos inusuales con modelos de una sola clase
Con este espacio de características comprimido en mano, el equipo entrena tres modelos de “una sola clase”: algoritmos diseñados para aprender la forma de un único grupo y marcar aquello que no encaja. Prueban Máquinas de Vectores de Soporte de Una Clase, Bosques de Aislamiento y un tipo de red neuronal llamada autoencoder. El autoencoder aprende a comprimir las características de cada péptido a una representación interna estrecha y luego reconstruirlas; los péptidos que pertenecen al patrón de enfermedad aprendido se reconstruyen con precisión, mientras que los inusuales acumulan un mayor error de reconstrucción. Comparar puntuaciones de anomalía normalizadas entre todos los métodos muestra que el autoencoder produce el conjunto más compacto de péptidos típicos y la separación más clara entre elementos internos y atípicos. Al fijar un umbral en el error de reconstrucción alrededor del percentil 95, el modelo clasifica a la mayoría de los péptidos como probablemente asociados a la enfermedad mientras marca de forma consistente una pequeña fracción como atípica.
Convertir puntuaciones complejas en un único número significativo
Para facilitar la interpretación biológica de los resultados, los autores introducen la Puntuación de Anomalía de Péptidos Asociados a la Enfermedad (DPAS). Esta puntuación combina dos ingredientes: cuán inusual parece un péptido para el autoencoder (su error de reconstrucción normalizado) y hasta qué punto sus características contribuyen a las predicciones, medido por un método popular de explicación llamado SHAP. En la práctica, los motivos y rasgos físico‑químicos específicos emergen como especialmente informativos. DPAS combina estas señales de modo que los péptidos que son a la vez estructuralmente extraños y respaldados por características biológicamente relevantes obtienen puntuaciones más altas. Los péptidos con mayor puntuación se examinan luego con una herramienta de búsqueda de motivos que los enlaza con firmas funcionales conocidas, como sitios de fosforilación, regiones de unión a metales y otros patrones regulatorios comunes en señalización y control enzimático.
Qué significa esto para futuros diagnósticos y fármacos
En términos prácticos, este trabajo ofrece un filtro más inteligente para encontrar péptidos sospechosos sin pretender que sabemos cuáles son definitivamente inofensivos. Al aprender únicamente de ejemplos confirmados relacionados con la enfermedad y luego priorizar nuevos candidatos con DPAS, los investigadores pueden seleccionar una lista breve y biológicamente plausible de péptidos para pruebas de laboratorio. Muchos de los candidatos mejor clasificados contienen motivos funcionales bien conocidos, lo que refuerza la idea de que pueden tener roles en procesos patológicos. Aunque el método aún depende de supuestos y carece de péptidos "seguros" validados experimentalmente para una validación completa, proporciona una base más realista y transparente para el descubrimiento de biomarcadores peptídicos y podría adaptarse a otros tipos de datos biológicos donde los ejemplos negativos fiables son escasos.
Cita: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Palabras clave: péptidos asociados a la enfermedad, detección de anomalías, autoencoder, descubrimiento de biomarcadores, aprendizaje de una sola clase