Clear Sky Science · es
El aprendizaje automático interpretable racionaliza la inhibición de la anhidrasa carbónica mediante predicción conformal y explicaciones contrafactuales
Por qué importan fármacos contra el cáncer más inteligentes
Los fármacos contra el cáncer a menudo actúan como herramientas toscas: aunque atacan las células tumorales, también pueden afectar a tejidos sanos y causar efectos secundarios graves. Una vía prometedora para afinar este objetivo es bloquear versiones específicas de una enzima llamada anhidrasa carbónica, que ayuda a los tumores a sobrevivir en entornos con poco oxígeno. Sin embargo, varias isoformas de esta enzima se parecen mucho entre sí, lo que dificulta diseñar medicamentos que ataquen las variantes “malas” en los tumores sin alterar la isoforma “buena” presente en todo el organismo. Este estudio muestra cómo el aprendizaje automático interpretable puede ayudar a los investigadores a afrontar este reto y diseñar candidatos a fármacos más selectivos y seguros.
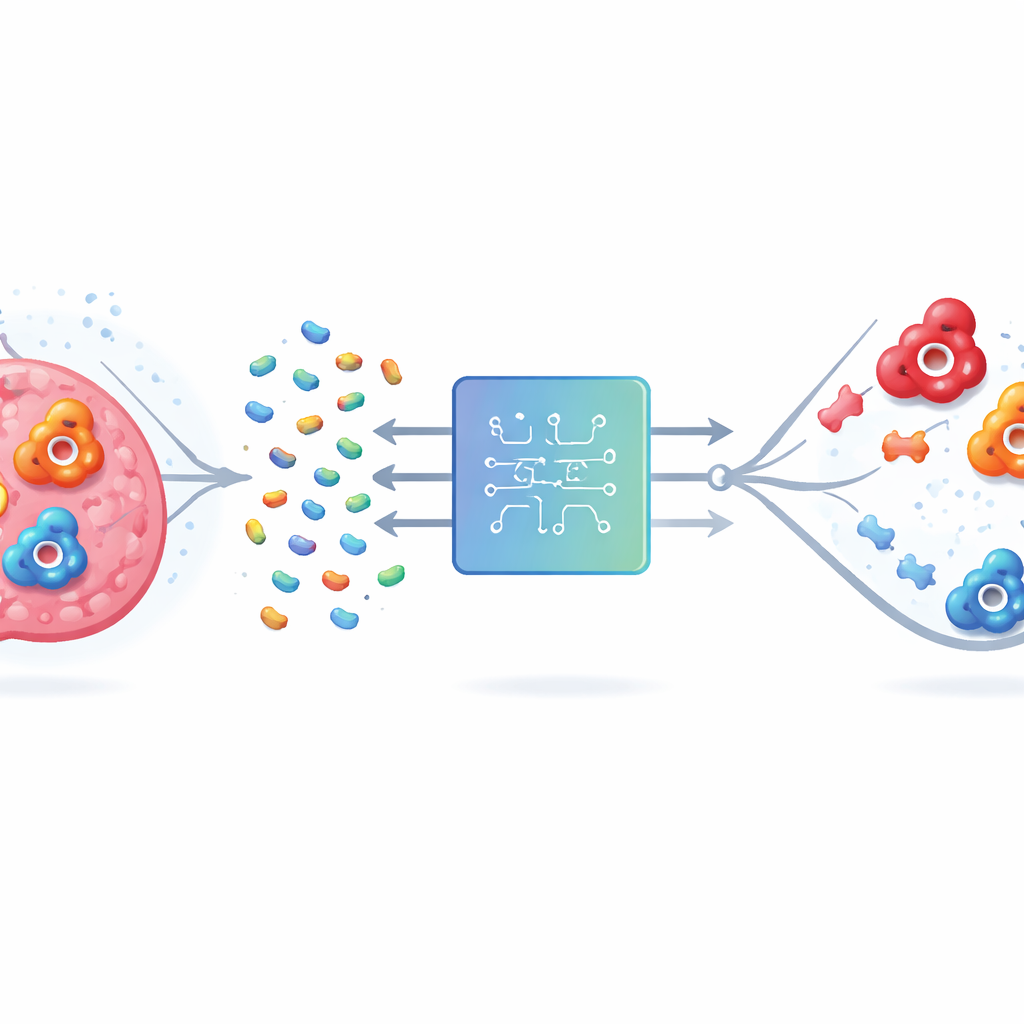
El problema de golpear el blanco equivocado
La anhidrasa carbónica humana (hCA) existe en muchas formas, o isoformas. Dos de ellas, IX y XII, están relacionadas con la supervivencia de las células cancerosas en tumores con falta de oxígeno, por lo que bloquearlas podría frenar la enfermedad y mejorar el tratamiento. Pero la isoforma II está muy extendida en tejidos sanos y tiene un sitio activo que se parece mucho al de IX y XII. Los fármacos que se unen a las tres pueden provocar problemas no deseados como acidosis metabólica y alteraciones visuales. Los métodos tradicionales de laboratorio y computacionales tienen dificultades porque las enzimas son moléculas grandes y complejas, y el número de compuestos con características de fármaco posibles es astronómicamente alto. Probarlos todos, ya sea en el laboratorio o por ordenador, no es factible.
Construir una base de datos limpia y de confianza
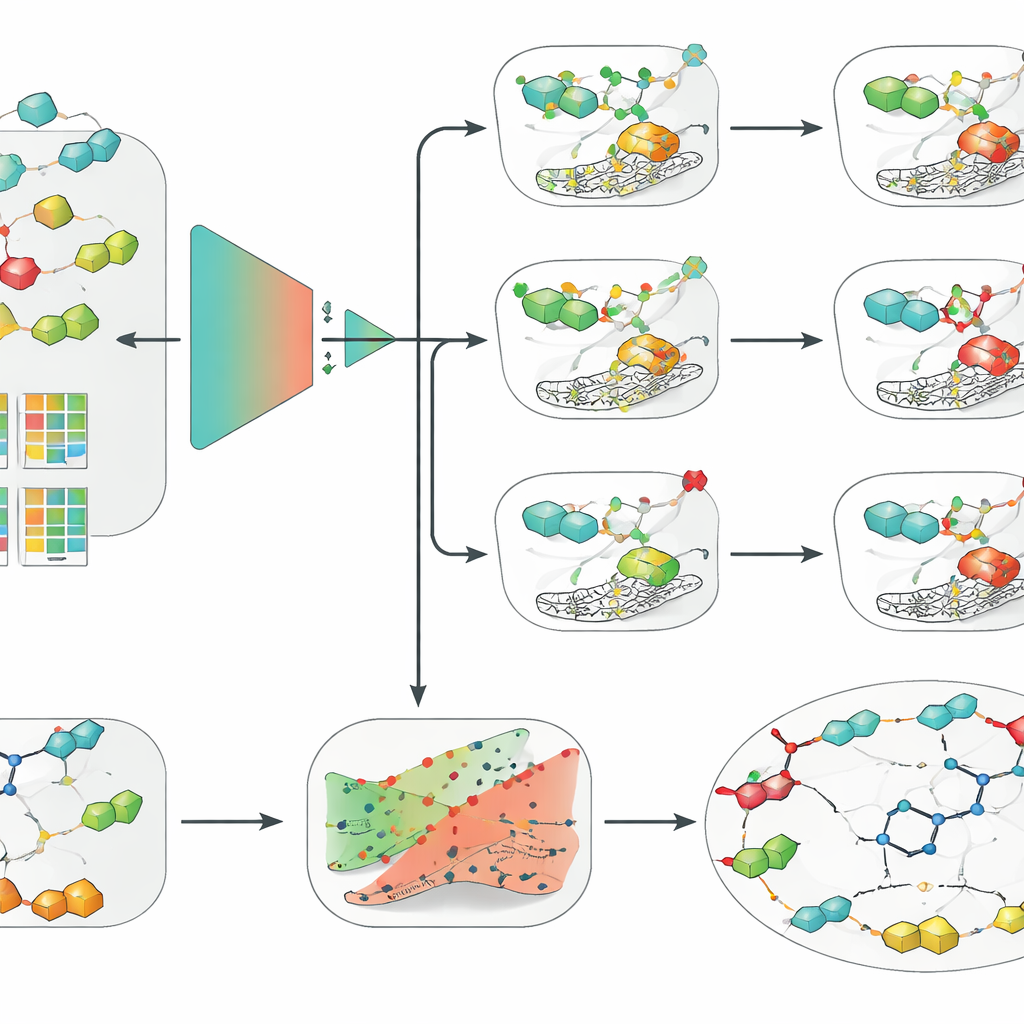
Los autores abordaron esto reuniendo primero una base de datos cuidadosamente depurada de miles de moléculas evaluadas frente a hCA II, IX y XII a partir del repositorio ChEMBL. Estandarizaron las estructuras químicas, eliminaron mediciones dudosas y se centraron en compuestos que comparten un grupo de unión al zinc común en esta clase de inhibidores. Usando umbrales estrictos, etiquetaron las moléculas como claramente activas o claramente inactivas y descartaron casos fronterizos que pudieran confundir a los modelos. Como había muchas más moléculas inactivas que activas, equilibraron los datos para que los algoritmos de aprendizaje no favorecieran simplemente a la clase mayoritaria. También usaron una división basada en andamiajes (“scaffold-based”) de los datos para que los conjuntos de entrenamiento y prueba contuvieran marcos moleculares centrales distintos, ofreciendo una imagen más realista de cómo los modelos manejarían compuestos verdaderamente nuevos.
Modelos simples superan al aprendizaje profundo cuando los datos son limitados
Con este conjunto de datos curado, el equipo comparó una amplia gama de enfoques, desde métodos clásicos de aprendizaje automático como regresión logística, bosques aleatorios y máquinas de vectores de soporte (SVM) hasta redes neuronales profundas modernas, incluidos modelos basados en grafos que operan directamente sobre las estructuras moleculares. Los emparejaron con varias formas de codificar moléculas, como descriptores tradicionales hechos a mano, huellas basadas en claves y representaciones aprendidas por un modelo de lenguaje químico. En las tres isoformas en estudio y bajo la evaluación más estricta basada en andamiajes, una combinación destacó de forma consistente: una SVM alimentada con huellas de conectividad extendida, una forma estructurada de describir los entornos químicos locales dentro de una molécula. Sorprendentemente, esta configuración comparativamente simple superó a modelos de grafos y de aprendizaje profundo más de moda, subrayando que la calidad de los datos, la validación cuidadosa y buenos descriptores moleculares pueden importar más que la complejidad algorítmica cuando los conjuntos de datos son de tamaño modesto.
Añadir confianza fiable y explicaciones amigables para el humano
Los investigadores envolvieron entonces su mejor modelo SVM en dos capas adicionales diseñadas para hacer sus predicciones más utilizables en el descubrimiento real de fármacos. Primero, aplicaron un marco llamado predicción conformal, que no solo entrega una respuesta binaria sino que proporciona una región de resultados probables junto con una tasa de error garantizada. Esto permite a los científicos ajustar cuán cautelosos quieren ser y reconocer casos en los que el modelo está genuinamente incierto. Segundo, usaron explicaciones contrafactuales para hacer el razonamiento del modelo más intuitivo. Para una molécula dada, generaron análogos estrechamente relacionados que invierten el resultado predicho de activo a inactivo, o viceversa. Al examinar estos pares para el candidato clínico SLC-0111, que bloquea selectivamente IX y XII pero no II, el método redescubrió de forma independiente una idea importante de la química medicinal: pequeños cambios en la “cola” de la molécula alteran fuertemente a qué isoforma prefiere unirse.
De los algoritmos a herramientas prácticas de diseño de fármacos
Para hacer su enfoque accesible, los autores empaquetaron los tres modelos SVM, la capa de incertidumbre y el motor de contrafactuales en una herramienta gráfica llamada CAInsight. Un usuario puede proporcionar la representación textual de una molécula y, con un solo clic, obtener la actividad predicha frente a hCA II, IX y XII, una estimación de cuán confiable es cada predicción y sugerencias de modificaciones estructurales que podrían aumentar o reducir la actividad. Aunque los modelos se centran en clasificar moléculas como activas o inactivas en lugar de predecir potencia exacta o selectividad en un solo paso, ya reproducen el comportamiento conocido de candidatos farmacológicos reales y distinguen cambios estructurales sutiles. Los autores señalan que conjuntos de datos más grandes y homogéneos, además de un análisis más profundo sobre cómo se eligen los umbrales de actividad, podrían afinar aún más el rendimiento.
Qué significa esto para futuros fármacos contra el cáncer
En términos sencillos, este trabajo demuestra que modelos de aprendizaje automático construidos con cuidado y bien explicados pueden ayudar a los químicos a diseñar fármacos contra el cáncer que distingan mejor entre blancos enzimáticos parecidos. Al combinar estadísticas robustas, estimaciones de incertidumbre y ejemplos intuitivos de “qué pasaría si”, el marco no solo predice qué moléculas probablemente funcionen, sino que también sugiere por qué. Este tipo de inteligencia artificial transparente podría acelerar el cribado virtual, apoyar el diseño generativo de nuevos compuestos y reducir la carga de ensayo y error en el laboratorio, ayudando en última instancia al descubrimiento de tratamientos más selectivos y seguros para los pacientes.
Cita: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Palabras clave: inhibidores de la anhidrasa carbónica, aprendizaje automático interpretable, selectividad de fármacos, predicción conformal, explicaciones contrafactuales