Clear Sky Science · es
Mutaciones compuestas heterocigotas en el gen CHAT, una de cambio de sentido y una variante en el sitio de empalme, en dos hermanos con síndrome miasténico congénito
Cuando la respiración falla sin aviso
Algunos niños parecen sanos al nacer y, sin embargo, dejan de respirar de forma súbita durante fiebres leves, necesitando ventilación de emergencia. Para sus familias, los episodios son aterradores y enigmáticos. Este estudio investiga a dos hermanos de Japón con episodios potencialmente mortales de debilidad y apnea (pausas en la respiración) y rastrea esas crisis hasta cambios minúsculos en un solo gen que ayuda a las neuronas a comunicarse con los músculos. Al combinar pistas clínicas, secuenciación genética y modelado proteico por ordenador, los investigadores muestran cómo estas mutaciones probablemente alteran una enzima clave y ofrecen a los médicos un objetivo más claro para el diagnóstico y el tratamiento.
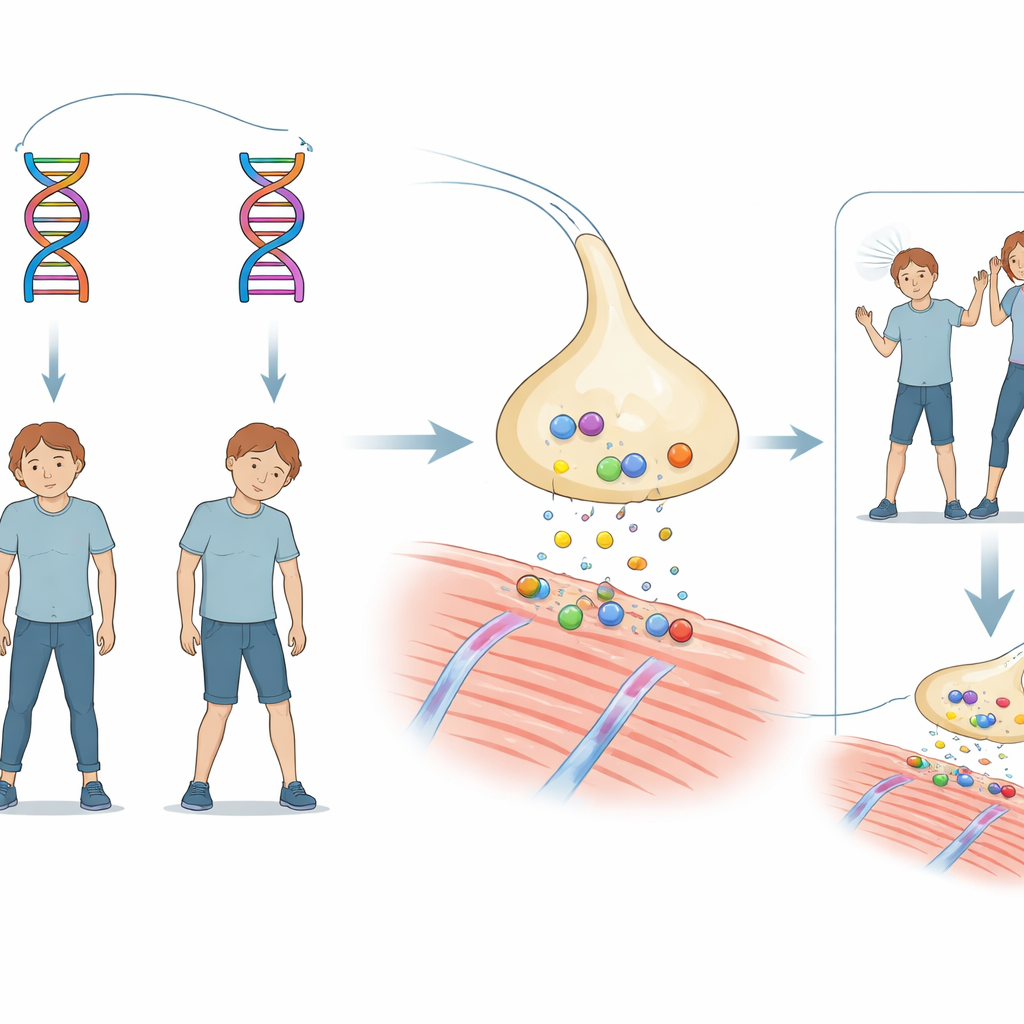
Un misterio familiar de debilidad súbita
La historia se centra en un hermano y una hermana que ambos tuvieron un desarrollo motor algo lento en la infancia. Alrededor de los 18 meses, cada uno experimentó episodios de apnea y pérdida de conciencia durante fiebres, lo bastante graves como para requerir ventilación mecánica. Conforme crecieron, ambos continuaron sufriendo episodios de ptosis palpebral y debilidad muscular generalizada desencadenados por infecciones, fiebre o esfuerzo. Las exploraciones cerebrales fueron normales y se descartaron las formas más comunes de miastenia mediadas por anticuerpos. Sin embargo, un fármaco que potencia la señal química entre nervio y músculo mejoró claramente sus síntomas, lo que apuntó a una rara enfermedad hereditaria llamada síndrome miasténico congénito.
Encontrando las instrucciones defectuosas
Para buscar una causa heredada, el equipo secuenció todos los genes codificantes de proteínas en los hermanos y sus progenitores. Encontraron que cada niño portaba dos alteraciones diferentes en el mismo gen, CHAT, que codifica la colina acetiltransferasa—una enzima que sintetiza acetilcolina, el principal mensajero químico que usan las neuronas para activar los músculos. Una de las alteraciones modificaba un único bloque constituyente de la enzima (una mutación de cambio de sentido conocida como G411R). La otra se situaba en un límite crítico donde la célula normalmente corta y une segmentos del gen al formar el ARN (una mutación en el sitio de empalme etiquetada c.752+2T>C). Cada progenitor llevaba solo una de estas variantes y estaba sano; únicamente los hijos que heredaron ambas manifestaron la enfermedad, lo que sugiere que el par de mutaciones en conjunto debilita la función enzimática.
Indagando cómo un corte oculto altera la enzima
Dado que los investigadores no pudieron obtener suficiente ARN natural de CHAT a partir de células sanguíneas, utilizaron un experimento de “minigen”. Clonaron el tramo relevante del gen en un vector de ADN, introdujeron la versión normal o la mutada en células cultivadas y luego examinaron cómo se procesaba el ARN. En el constructo normal, el ARN contenía todos los segmentos esperados. En la versión mutante, se omitió por completo un segmento conocido como exón 5, aunque el marco de lectura global del gen permaneció intacto. Esto significaba que la enzima se produciría pero carecería de un breve tramo interno de aminoácidos. Comparaciones evolutivas mostraron que esta región ausente está muy conservada entre especies, lo que sugiere que desempeña un papel estructural importante.
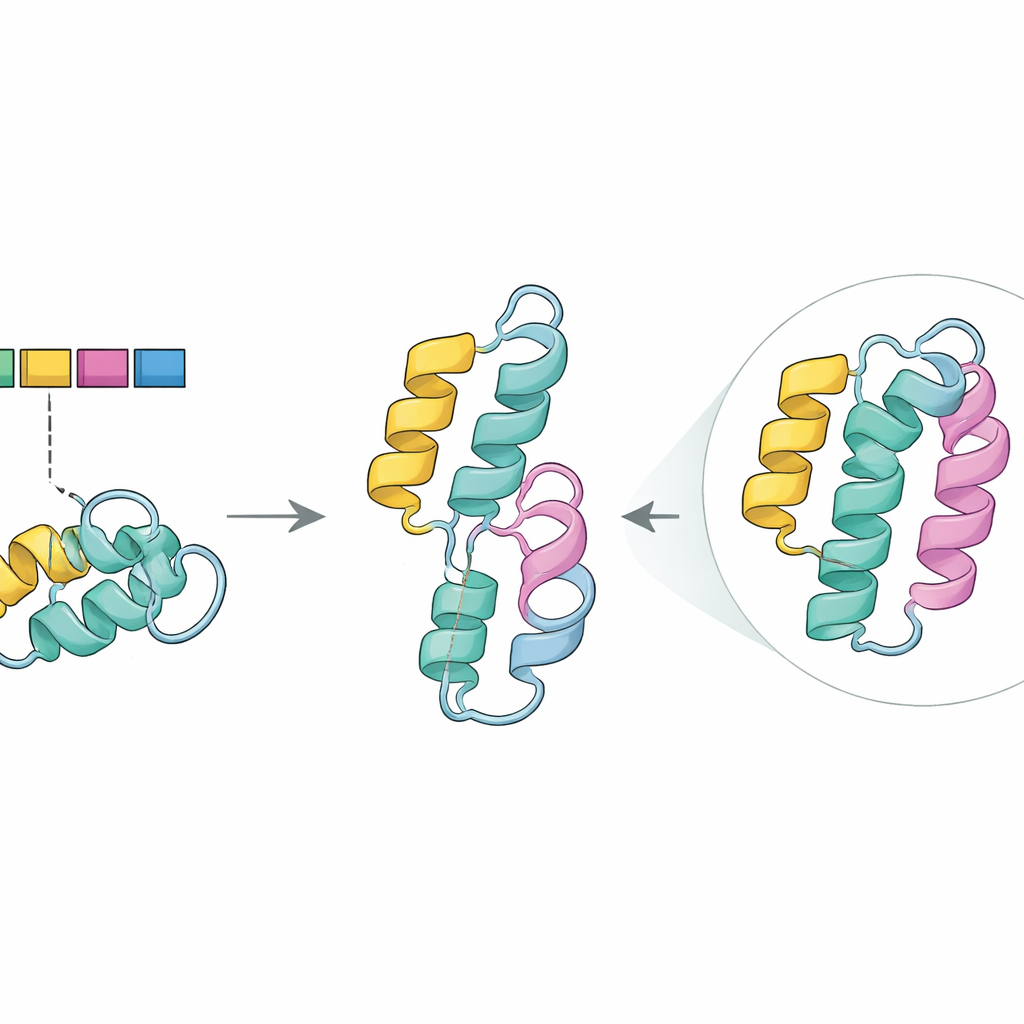
Observando el daño estructural in silico
Para explorar ese papel, el equipo recurrió a AlphaFold2, un programa avanzado que predice formas tridimensionales de proteínas a partir de sus secuencias. En la enzima normal, la porción codificada por el exón 5 forma una de las espirales compactas (una hélice alfa) que ayudan a estabilizar el núcleo de la proteína. En la estructura mutante predicha, esa hélice desaparecía, dejando un hueco en una región conocida por trabajos previos como crucial para mantener la estabilidad y favorecer una química eficiente. Junto con herramientas informáticas que señalan mutaciones dañinas, estos resultados respaldan la idea de que la omisión del exón 5, especialmente cuando se combina con el cambio G411R en la otra copia del gen, socava el rendimiento de la enzima sin eliminarlo por completo—coherente con los síntomas moderados pero graves observados en los hermanos.
Qué significa esto para pacientes y familias
El estudio concluye que la combinación de la mutación de cambio de sentido G411R y la variante recientemente identificada en el sitio de empalme de CHAT es muy probablemente responsable del síndrome miasténico congénito de los hermanos. Al demostrar, mediante el ensayo de minigen y el modelado estructural, cómo la alteración del sitio de empalme elimina una hélice estabilizadora de la enzima, los autores ofrecen una explicación mecanicista sobre la que clínicos e investigadores pueden avanzar. Para las familias afectadas, este trabajo aporta más que un nombre: respalda tratamientos ajustados con fármacos que potencian la señal neuromuscular, orienta el asesoramiento genético para futuros embarazos y añade un ejemplo importante a un catálogo de cómo cambios sutiles en nuestro código genético pueden influir profundamente en la fuerza muscular y en el acto esencial de respirar.
Cita: Kikuchi, S., Wada, N., Mariya, T. et al. Compound heterozygous CHAT gene mutations, a missense and a splice site variant, in two siblings with congenital myasthenic syndrome. Sci Rep 16, 9346 (2026). https://doi.org/10.1038/s41598-026-39759-y
Palabras clave: síndrome miasténico congénito, gen CHAT, colina acetiltransferasa, mutación en el sitio de empalme, unión neuromuscular