Clear Sky Science · es
La deconvolución mejorada guiada por atención permite estimar tipos celulares sin referencia en transcriptómica espacial
Ver las células en su lugar
La biología moderna puede leer la actividad de miles de genes a la vez, no solo en células aisladas sino directamente dentro de cortes finos de tejido. Esta visión de «transcriptómica espacial» revela dónde viven e interactúan diferentes células, pero cada medida a menudo mezcla señales de muchas células vecinas. El estudio presenta un nuevo método computacional, llamado AGED, que puede desenredar estas mezclas y estimar qué tipos celulares están presentes en cada lugar, sin necesidad de un conjunto de referencia de células individuales separado y cuidadosamente emparejado. 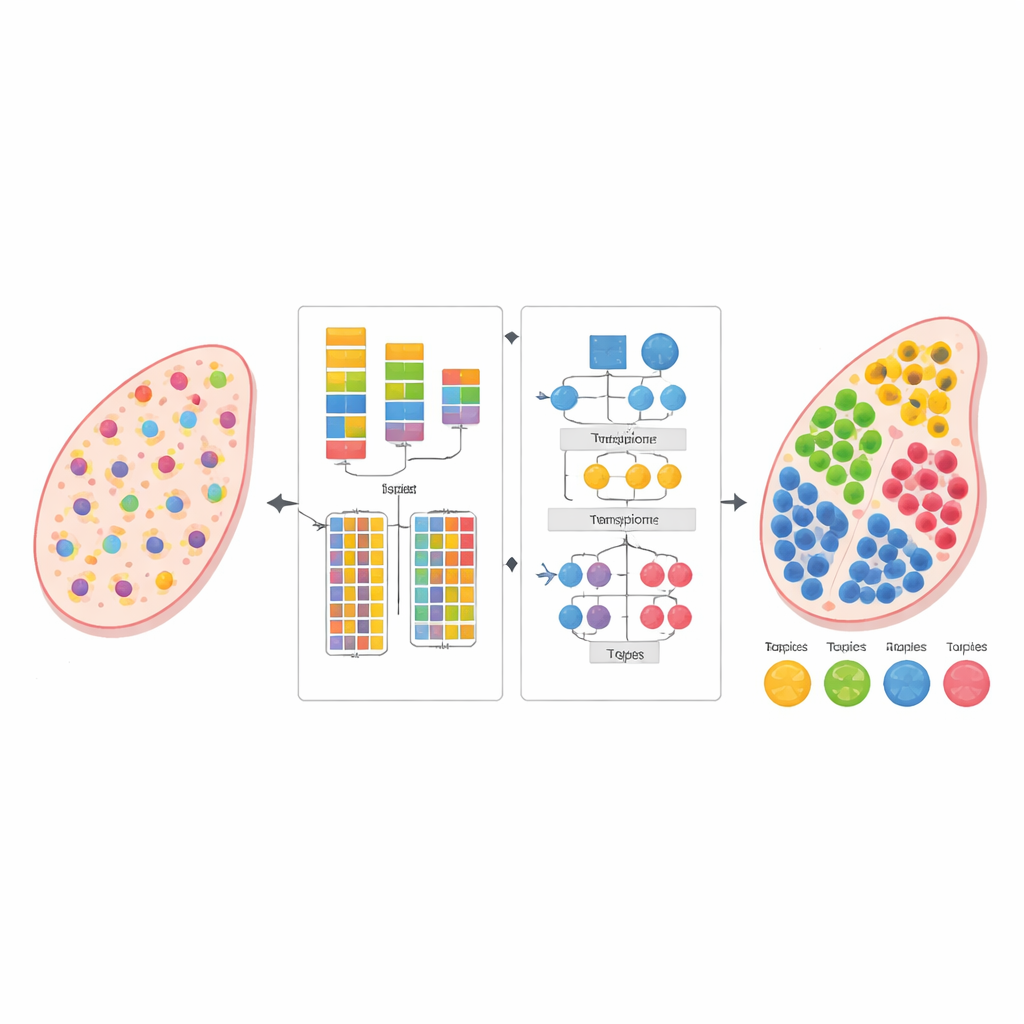
Por qué es difícil cartografiar células en tejidos
Las plataformas de transcriptómica espacial miden la actividad génica en una cuadrícula de puntos sobre un corte de tejido. Dado que la mayoría de los puntos capturan varias células a la vez, los investigadores deben descomponer matemáticamente las señales mezcladas para recuperar los tipos celulares subyacentes y sus proporciones. Las herramientas existentes a menudo dependen de atlas de referencia de células individuales del mismo tejido. Esos atlas pueden faltar para tejidos raros, estados patológicos especiales o condiciones experimentales inusuales, e incluso cuando existen pueden no coincidir perfectamente, introduciendo sesgos. Los métodos sin referencia evitan esta dependencia, pero los enfoques actuales tienen dificultades con patrones espaciales complejos, relaciones génicas sutiles y el reto de decidir cuántos tipos celulares distintos buscar en primer lugar.
Una estrategia en dos pasos para desenredar mezclas
Los autores diseñaron AGED como un marco en dos etapas que combina ideas de la estadística y del aprendizaje profundo moderno. En la primera etapa, el método prueba un rango de posibilidades sobre cuántos tipos celulares podrían estar presentes en el tejido. Utiliza una red neuronal basada en atención rápida, conocida como Performer, para aprender descomposiciones candidatas y luego las puntúa con varios criterios a la vez: qué tan bien el modelo reconstruye los recuentos génicos observados, qué tan claramente se separan los grupos celulares inferidos y qué tan diversos son esos grupos. Un procedimiento de ajuste de curvas encuentra un «punto de codo» donde añadir más tipos celulares aporta poco beneficio, lo que permite al método seleccionar automáticamente un número adecuado en lugar de depender de la estimación del usuario.
Atención guiada para capturar la biología
Una vez fijado el número de tipos celulares, la segunda etapa de AGED refina la solución con una arquitectura de atención más rica. Parte de un modelo estadístico de temas que trata cada punto del tejido como una mezcla de «temas» ocultos —aquí en lugar de los tipos celulares— y cada tipo celular como un patrón génico característico. Estos temas iniciales proporcionan estructura global. El modelo luego superpone varios mecanismos de atención: uno conecta los temas estadísticos con la red neuronal, otro agrupa información de puntos vecinos en el espacio físico y un tercero vincula directamente los temas con los genes. Un sistema de compuertas permite al modelo decidir, en cada caso, cuánto confiar en los patrones estadísticos previos frente a los datos locales. Restricciones adicionales fomentan soluciones dispersas, reflejando la realidad biológica de que la mayoría de las ubicaciones tisulares están dominadas por solo unos pocos tipos celulares principales. 
Poniendo el método a prueba
Los investigadores evaluaron AGED en varios tipos de datos. En tejido simulado del bulbo olfatorio de ratón, el método recuperó cuatro capas anatómicas conocidas y coincidió con las composiciones celulares verdaderas más estrechamente que herramientas de referencia y sin referencia ampliamente usadas, logrando tanto una alta correlación con la verdad de referencia como un bajo error de reconstrucción. En adenocarcinoma ductal pancreático humano, AGED eligió automáticamente una solución de veinte tipos celulares que se alineó con regiones anotadas por patólogos, como tumor, conducto y páncreas normal, superando a otros métodos en una medida de similitud estructural que compara los mapas inferidos con la estructura tisular visible. En tejido de timo humano, AGED separó con precisión poblaciones celulares clave y capturó una relación negativa biológicamente esperada entre dos tipos celulares epiteliales especializados —un patrón que los enfoques competidores no consiguieron reproducir. Análisis adicionales en otros conjuntos de datos y a resolución similar a la unicelular apoyaron más la robustez del método.
Qué significa esto de cara al futuro
Para un no especialista, AGED puede verse como un motor inteligente de separación de mezclas para tejidos complejos: aprende cuántas comunidades celulares distintas están presentes, dónde se localizan y qué genes las definen, todo a partir de los propios datos espaciales. Al entrelazar modelos estadísticos interpretables con redes neuronales flexibles basadas en atención, el marco ofrece tanto precisión como comprensión, incluso cuando no existe un atlas de referencia adecuado. Esto lo convierte en una herramienta práctica para explorar la organización tisular en salud y enfermedad, desde capas cerebrales hasta tumores y órganos inmunes, y apunta hacia una estrategia más amplia para usar conocimiento previo que guíe modelos de aprendizaje automático potentes pero opacos en biología.
Cita: Yang, X., Wang, Y. & Chen, X. Attention-guided enhanced deconvolution enables reference-free cell type estimation in spatial transcriptomics. Sci Rep 16, 8097 (2026). https://doi.org/10.1038/s41598-026-39703-0
Palabras clave: transcriptómica espacial, deconvolución de tipos celulares, aprendizaje profundo, arquitectura tisular, análisis sin referencia