Clear Sky Science · es
Modelado matemático y cálculo de índices NM‑polinomiales para la predicción de propiedades fisicoquímicas
Por qué esto importa para los medicamentos del futuro
Diseñar un nuevo medicamento es algo parecido a diseñar una aeronave: se quiere conocer su comportamiento mucho antes de construirla en la realidad. En el caso de los fármacos, ese comportamiento incluye con qué facilidad se evaporan, cómo se mezclan con agua o con grasas y cómo se desplazan por el organismo. Este artículo muestra cómo matemáticas cuidadosamente formuladas pueden predecir muchas de estas características físicas y químicas a partir de la estructura de un fármaco, lo que puede ahorrar tiempo, costes y ensayo y error en el descubrimiento de fármacos. 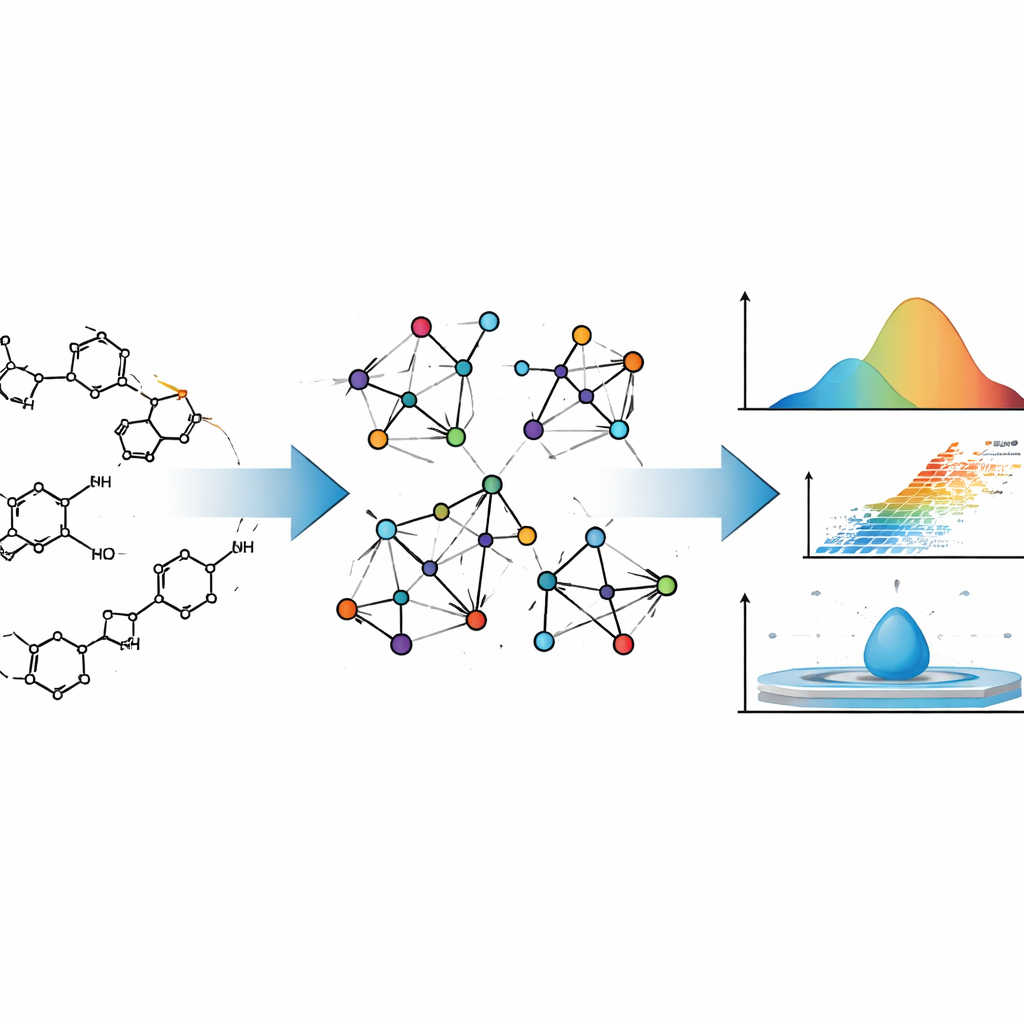
De las moléculas a las redes
Los autores tratan las moléculas de los fármacos no solo como colecciones de átomos, sino como redes. En esta representación, cada átomo es un punto y cada enlace químico es una línea que conecta dos puntos. Este tipo de descripción proviene de la teoría de grafos, una rama de las matemáticas que estudia redes de todo tipo, desde enlaces en redes sociales hasta redes eléctricas. Los químicos han usado estos “grafos moleculares” durante décadas, porque ciertos resúmenes numéricos de estos grafos —llamados índices topológicos— a menudo reflejan cómo se comportan las moléculas en el mundo real, por ejemplo cuán fácilmente hierven o cuán densas son.
Añadiendo detalle de vecindad a la imagen
Los índices tradicionales suelen fijarse únicamente en cuántos enlaces toca cada átomo. El equipo detrás de este estudio va un paso más allá. Utilizan los llamados índices NM‑polinomiales (neighborhood M‑polynomial), que no solo cuentan las conexiones de un átomo sino que también resumen cuán conectados están sus vecinos. Esta descripción más rica captura sutilezas como cuán ramificada está una molécula, cómo se fusionan sus anillos y dónde se sitúan átomos de oxígeno o nitrógeno dentro del entramado. Esas características a su vez influyen en cómo se adhieren las moléculas entre sí, en su rigidez y en cómo responden sus electrones a campos eléctricos, todos ellos ingredientes de propiedades fisicoquímicas clave.
Probando la idea con fármacos anticáncer reales
Para anclar sus matemáticas en la realidad, los autores calculan primero índices NM‑polinomiales para dos conocidos agentes anticáncer, Mitoxantrona y Doxorrubicina. Ambos son moléculas complejas con múltiples anillos y se usan ampliamente en quimioterapia. Al traducir sus dibujos químicos detallados en grafos moleculares y luego en índices NM‑polinomiales, los autores muestran cómo el método rastrea sistemáticamente los cambios estructurales a lo largo de diferentes “tamaños” de estas moléculas. Después automatizan este proceso con un programa en Python, que toma la conectividad de una molécula (en forma de matriz de adyacencia) y devuelve al instante el conjunto completo de índices, minimizando el error humano y acelerando cálculos que serían tediosos a mano. 
Enseñando a las máquinas a leer huellas moleculares
A continuación, los investigadores amplían el estudio más allá de esos dos fármacos a una colección de 45 medicamentos policíclicos, incluidos nombres comunes como acetaminofeno, ibuprofeno y varias terapias dirigidas modernas. Para cada fármaco compilan nueve índices NM‑polinomiales y nueve propiedades medidas experimentalmente: complejidad, punto de ebullición, entalpía de vaporización, punto de inflamación, refractividad molar, polarizabilidad, tensión superficial, volumen molar e índice de refracción. Luego entrenan varios modelos de regresión al estilo de aprendizaje automático —Lineal, Ridge, Lasso y Elastic Net— para aprender cómo combinaciones de índices se traducen en cada propiedad. Se aplican salvaguardas estadísticas cuidadosas: eliminan entradas redundantes, estandarizan variables, realizan validación cruzada repetida sobre el 80% de los datos y prueban los modelos finales en un 20% no visto.
Lo que revelan los números
Los modelos muestran que los índices NM‑polinomiales son particularmente potentes para propiedades vinculadas a cómo se empaquetan e interactúan las moléculas. Para el punto de ebullición, entalpía de vaporización, punto de inflamación, refractividad molar, polarizabilidad y volumen molar, los mejores modelos alcanzan puntuaciones de correlación muy altas, lo que significa que los valores predichos siguen de cerca a los experimentales. Métodos regularizados como Ridge y Elastic Net suelen rendir mejor, lo que sugiere que imponer restricciones suaves ayuda a que los modelos se concentren en los aspectos más informativos de los índices. Un mapa de correlaciones confirma que varios índices —especialmente los relacionados con la conectividad global y la “riqueza de la vecindad”— se alinean de forma fuerte y consistente con estas propiedades en el panel de 45 fármacos.
Límites y margen de mejora
No todas las propiedades cooperan. El índice de refracción, que captura cómo se desvía la luz al entrar en un material, resulta problemático: los modelos tienen dificultades para superar la precisión de simples promedios y los índices NM‑polinomiales muestran solo correlaciones débiles con él. La tensión superficial se captura de forma moderada, pero no tan bien como otras características. Estas lagunas indican que algunos comportamientos dependen de rasgos más allá de la conectividad bidimensional, como la forma tridimensional o efectos electrónicos sutiles. Los autores sugieren que trabajos futuros podrían combinar índices NM‑polinomiales con descriptores cuántico‑químicos o 3D para salvar esa brecha.
Qué significa esto para el diseño de fármacos
En términos sencillos, el estudio demuestra que matemáticas sofisticadas pero bien estructuradas pueden convertir un esquema estático de una molécula en un predictor sorprendentemente preciso de cómo se comportará en laboratorio. Para muchas propiedades importantes —qué difícil es hervir, cuánto ocupa o con qué facilidad se desplazan sus electrones— el enfoque NM‑polinomial, combinado con técnicas de regresión modernas, iguala o supera a métodos anteriores que usaban índices más simples o conjuntos de datos menores. Aunque aún no sustituye por completo a los experimentos, ofrece a los diseñadores de fármacos una herramienta de cribado más rápida: calculando estas huellas basadas en grafos pueden estimar propiedades fisicoquímicas clave desde etapas tempranas, centrar el trabajo de laboratorio en los candidatos más prometedores y explorar el espacio químico con mayor eficiencia.
Cita: Tawhari, Q.M., Naeem, M., Koam, A.N.A. et al. Mathematical Modeling and Computation of NM-Polynomial Indices for Physicochemical Properties Prediction. Sci Rep 16, 8136 (2026). https://doi.org/10.1038/s41598-026-39562-9
Palabras clave: teoría de grafos químicos, predicción de propiedades de fármacos, topología molecular, aprendizaje automático en química, descriptores fisicoquímicos