Clear Sky Science · es
Modelo de aprendizaje analítico dirigido por genes para un diagnóstico preciso del cáncer de mama
Por qué esta investigación importa para pacientes y familias
El cáncer de mama es hoy el cáncer más diagnosticado en mujeres en todo el mundo, y pacientes que parecen tener la misma enfermedad en los papeles pueden presentar resultados muy distintos. Este estudio muestra cómo los patrones en miles de genes, combinados con un sistema de inteligencia artificial cuidadosamente diseñado, pueden ayudar a los médicos a detectar con mayor fiabilidad quién tiene cáncer y cuán grave puede ser—utilizando únicamente datos reales de pacientes y un conjunto compacto de genes clave.
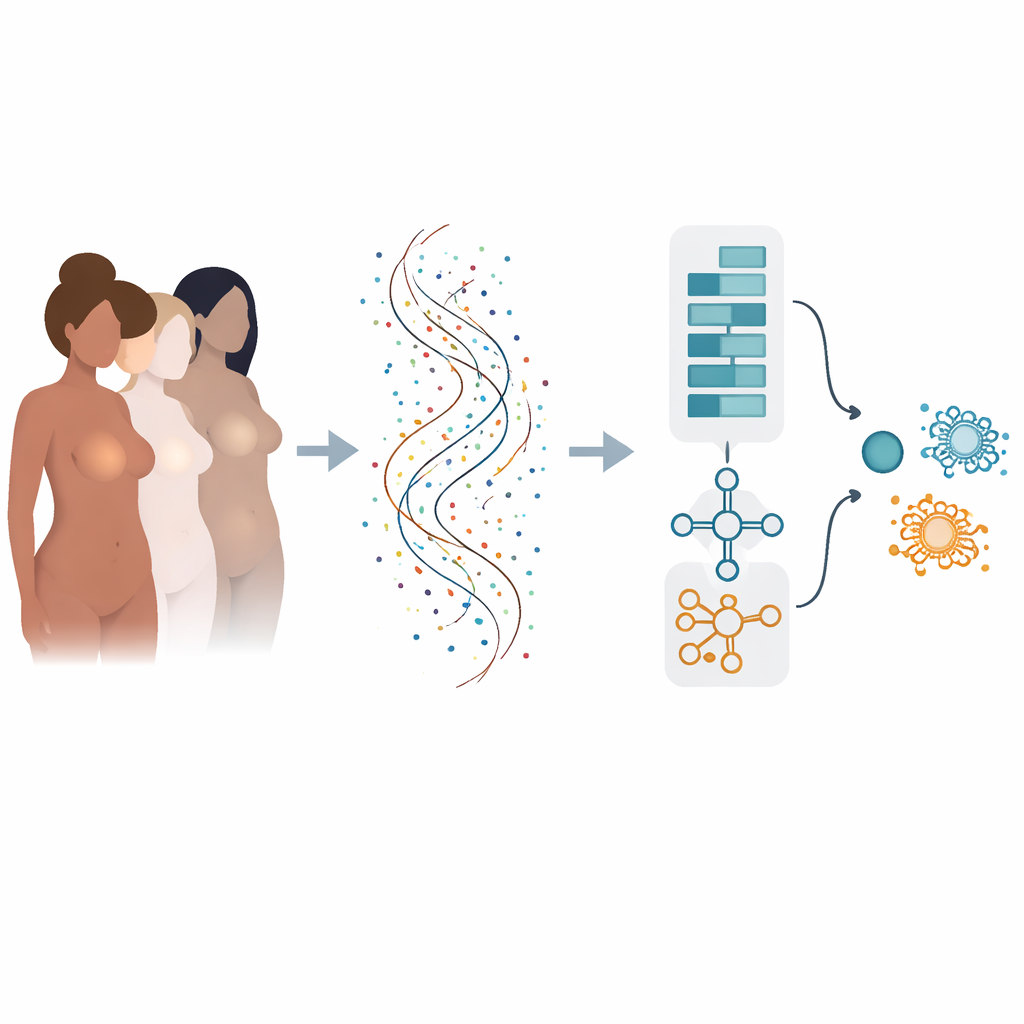
De muchos factores de riesgo al lenguaje de los genes
El riesgo de cáncer de mama está moldeado por muchas influencias: cambios genéticos heredados, hormonas, peso corporal, estilo de vida y más. Una vez que aparece el cáncer, su comportamiento viene determinado por qué genes están activados o desactivados dentro de cada tumor. La secuenciación moderna puede medir la actividad de decenas de miles de genes a la vez, pero convertir este océano de números en respuestas claras de sí o no para diagnóstico y pronóstico es difícil. Los métodos informáticos tradicionales a menudo analizan los genes uno a uno y pueden pasar por alto la forma en que grupos de genes actúan conjuntamente, o pueden funcionar bien solo en un conjunto de datos y fallar al probarse en otro lugar.
Enseñar a un modelo de doble cerebro a leer patrones génicos
Los autores construyeron un modelo híbrido de aprendizaje profundo que actúa un poco como dos cerebros especializados trabajando juntos. Una parte, inspirada en el análisis de imágenes, explora una lista ordenada de genes para detectar patrones locales—clústeres de genes cuya actividad conjunta señala la presencia de cáncer. La otra parte trata los mismos genes como una secuencia, aprendiendo cómo los genes “conductores” tempranos y los genes “descendientes” posteriores se influyen entre sí a lo largo de la lista. Al combinar estas dos perspectivas, el modelo puede capturar tanto relaciones de corto alcance como de largo alcance dentro de la huella genética del tumor.
Encontrar un núcleo estable de genes señal
En lugar de introducir las 17.815 genes medidos en el modelo, el equipo diseñó una canalización estricta y “libre de fugas” para seleccionar solo los más informativos. Usando una medida estándar de correlación dentro de bucles de verificación cruzada repetidos, clasificaron repetidamente los genes según la fuerza con la que su actividad se asociaba al estado canceroso. Luego mantuvieron únicamente los genes que consistentemente llegaron a la cima en todos los subconjuntos de entrenamiento, resultando en una firma estable de 236 genes. Los investigadores también mapearon cómo interactúan estos genes entre sí, mostrando que muchos forman redes fuertemente conectadas relacionadas con el crecimiento tumoral, el metabolismo, la inmunidad y el entorno tisular circundante—evidencia de que el conjunto elegido refleja biología real, no ruido aleatorio.
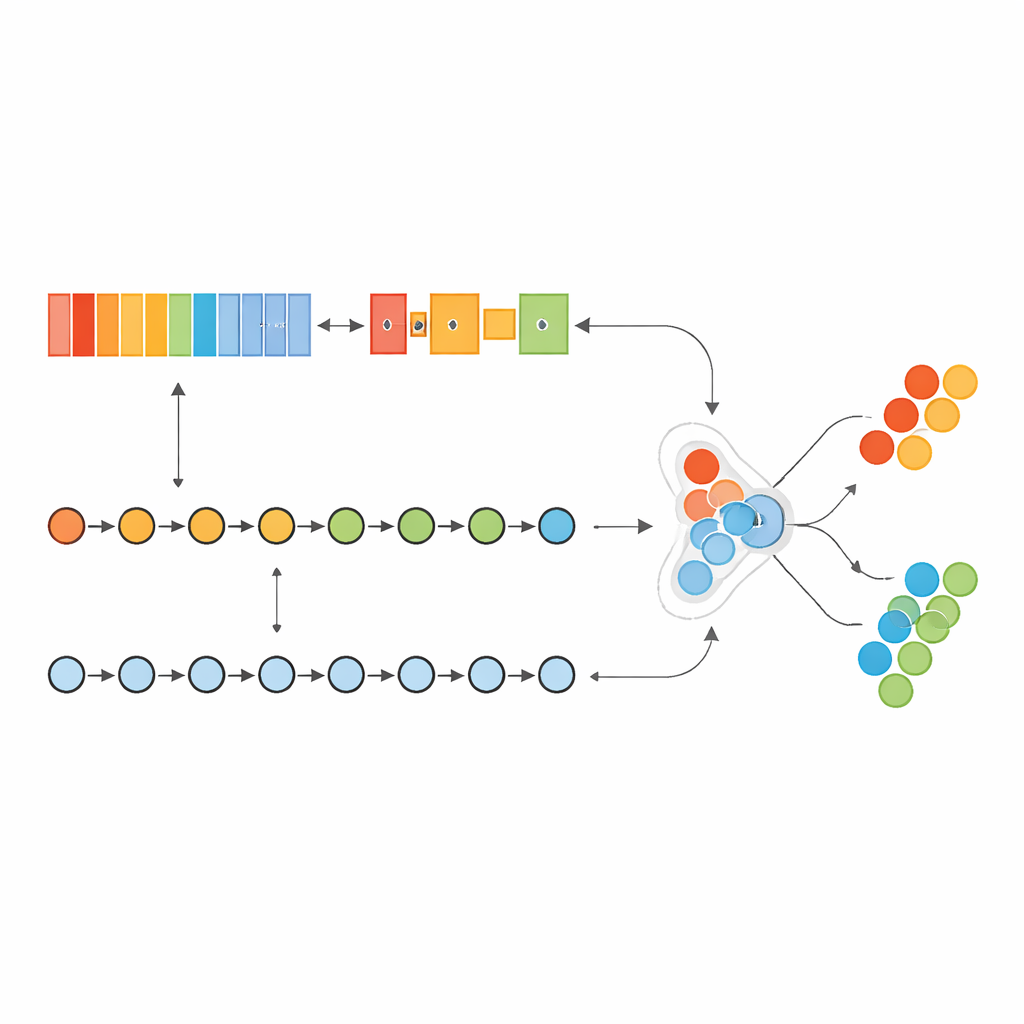
Poner el modelo a prueba
El sistema híbrido se entrenó y ajustó con muestras de cáncer de mama del Cancer Genome Atlas y luego se desafió con un conjunto de datos completamente separado conocido como METABRIC. Para manejar el hecho de que las muestras cancerosas superan con creces a las normales, los autores no crearon datos artificiales; en su lugar, ajustaron cuánto “importa” al modelo equivocarse en la clase menos frecuente. Tras una búsqueda automatizada de los mejores parámetros, el modelo alcanzó puntuaciones cercanas a la perfección en su conjunto de datos principal, marcando correctamente casi todos los casos de cáncer y generando prácticamente ninguna alarma falsa. Importante, el rendimiento se mantuvo extremadamente alto y muy estable incluso cuando el modelo se aplicó a la cohorte externa METABRIC, lo que sugiere que el enfoque puede generalizar más allá de un solo estudio u hospital.
Qué significa esto para la atención futura
En términos sencillos, este trabajo entrega una IA afinada en dos partes que lee un código compacto de 236 genes para distinguir muestras mamarias cancerosas de no cancerosas con notable precisión y consistencia, incluso en condiciones ruidosas. Aunque el estudio actual solo analiza la actividad génica y usa datos de pacientes pasados, sus métodos sientan las bases para futuras herramientas que podrían combinar múltiples tipos de datos—como imágenes tisulares y capas moleculares adicionales—y proporcionar explicaciones claras de qué genes impulsan cada predicción. Con una validación adicional en estudios clínicos prospectivos, un sistema así podría convertirse en una columna vertebral universal para el diagnóstico de precisión del cáncer de mama, ayudando a los médicos a personalizar el tratamiento usando la “firma” genética del tumor de cada paciente.
Cita: Hesham, F., Abbassy, M.M. & Abdalla, M. Gene driven analytical learning model for accurate breast cancer diagnosis. Sci Rep 16, 8155 (2026). https://doi.org/10.1038/s41598-026-39430-6
Palabras clave: diagnóstico del cáncer de mama, expresión génica, aprendizaje profundo, CNN-BiLSTM, oncología de precisión