Clear Sky Science · es
CiCLoDS: Agrupación conjunta de células y selección de genes para transcriptómica espacial unicelular
Encontrando vecindarios en la ciudad de las células
Los microscopios modernos pueden ahora determinar qué genes están activos en cientos de miles de células manteniendo a cada célula en su lugar original dentro del tejido. Esta revolución de la “transcriptómica espacial” es como convertir un mapa borroso de la ciudad en una vista a nivel de calle de cada casa. Pero hay un inconveniente: estos mapas contienen mediciones de miles de genes por célula, mucho más de lo que los científicos pueden interpretar fácilmente o permitirse medir en experimentos posteriores. Este estudio presenta CiCLoDS, un nuevo método que encuentra vecindarios celulares significativos y, al mismo tiempo, selecciona una lista pequeña e interpretable de genes que definen esos vecindarios.
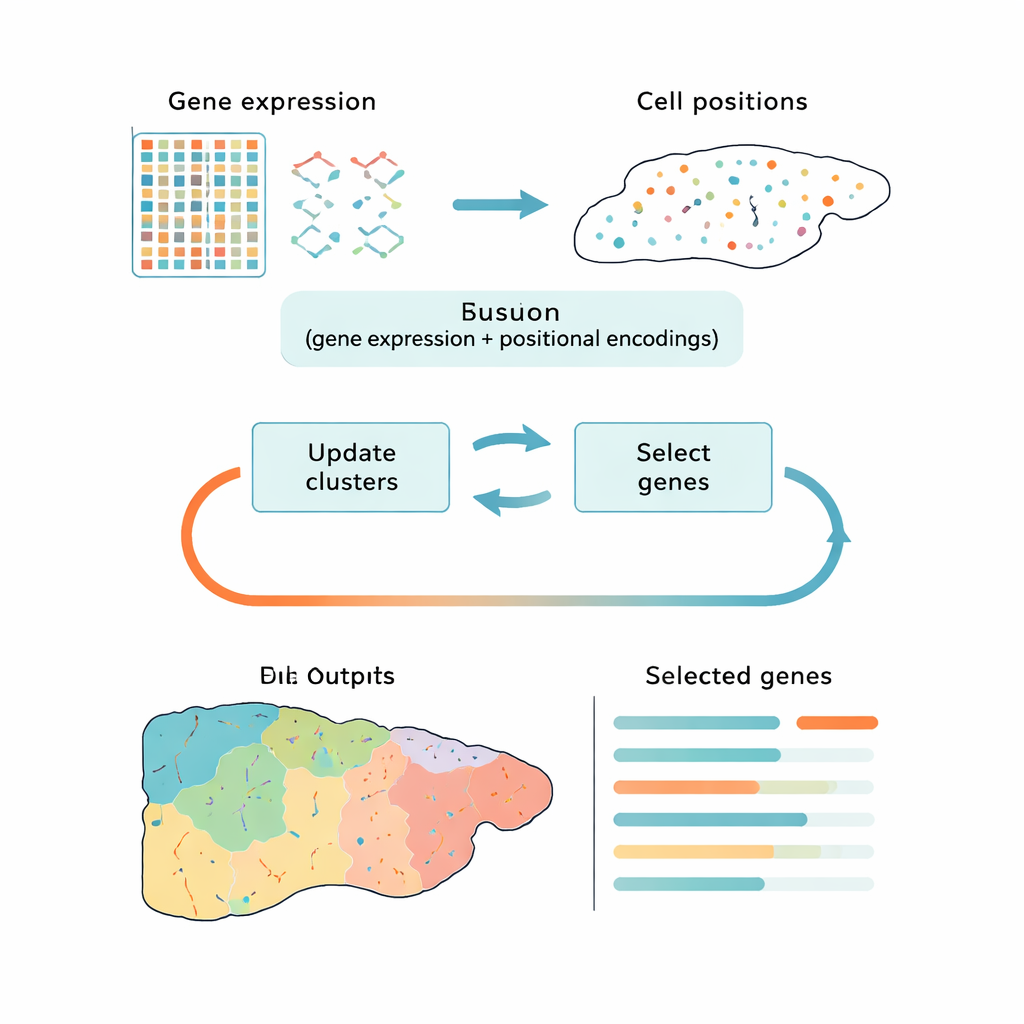
Una forma más inteligente de reducir grandes datos
La mayoría de las herramientas actuales abordan este desafío en dos pasos desconectados: primero reducen los datos a una forma más simple y luego agrupan las células en clústeres. Enfoques populares como el análisis de componentes principales (PCA) preservan la variación global pero pueden centrarse en ruido técnico o señales genéricas del ciclo celular en lugar de las diferencias biológicas que importan. Otros métodos usan aprendizaje profundo para encontrar patrones, pero actúan como cajas negras y no indican claramente qué genes son más importantes. CiCLoDS toma una ruta diferente. Trata la selección de genes y la agrupación como un único problema conjunto bajo un “presupuesto” definido por el usuario sobre cuántos genes se pueden conservar. En efecto, plantea la pregunta: ¿qué conjunto limitado de genes explica mejor cómo las células se distribuyen en grupos distintos, considerando tanto su actividad génica como, cuando está disponible, su posición física en el tejido?
De las matemáticas a los mapas de tejidos reales
Los autores adaptan una familia de técnicas matemáticamente transparentes llamadas agrupación por subespacios a las realidades de la transcriptómica espacial, donde los conjuntos de datos pueden contener más de un millón de células. CiCLoDS funciona sobre una tabla simple de células por genes, asignando células a clústeres mientras puntúa cada gen según cuánto contribuye a separar esos clústeres. También puede integrar información espacial añadiendo “codificaciones” posicionales que describen dónde se sitúa cada célula en el tejido, sin cambiar la optimización central. En conjuntos de datos grandes de hígado de ratón y colon humano generados por plataformas de imagen de alta resolución, CiCLoDS se ejecuta en minutos en ordenadores estándar y produce paneles de genes compactos—del orden de unas pocas decenas a unos pocos cientos de genes—que aun así capturan la rica estructura de los datos originales.
Revelando zonas ocultas y vasos sanguíneos
Al aplicar CiCLoDS al hígado de ratón, el equipo preguntó si el método podía recuperar patrones conocidos de “zonación”: desplazamientos graduales en la función de los hepatocitos de un lado del lobulillo al otro. En comparación con PCA y una herramienta líder de selección génica llamada geneBasis, CiCLoDS produjo zonas espaciales más limpias con fronteras más definidas y muchas menos regiones mal asignadas, como se observa en métricas cuantitativas que miden la concordancia con un mapa de referencia. De manera notable, cuando se le permitió usar más genes, CiCLoDS redescubrió grupos de hepatocitos similares a periportal y pericentral que coincidían estrechamente con clústeres de referencia definidos por expertos, aun cuando no se le indicó el gen biomarcador clave AXIN2 ni se le proporcionaron coordenadas espaciales explícitas. Al añadir codificaciones espaciales, CiCLoDS también aprendió paneles de genes enriquecidos en funciones relacionadas con la superficie celular y los vasos, y pudo distinguir con precisión vasos sanguíneos reales de artefactos de imagen—algo que los métodos más simples no lograban o solo conseguían con ajustes ad hoc adicionales.
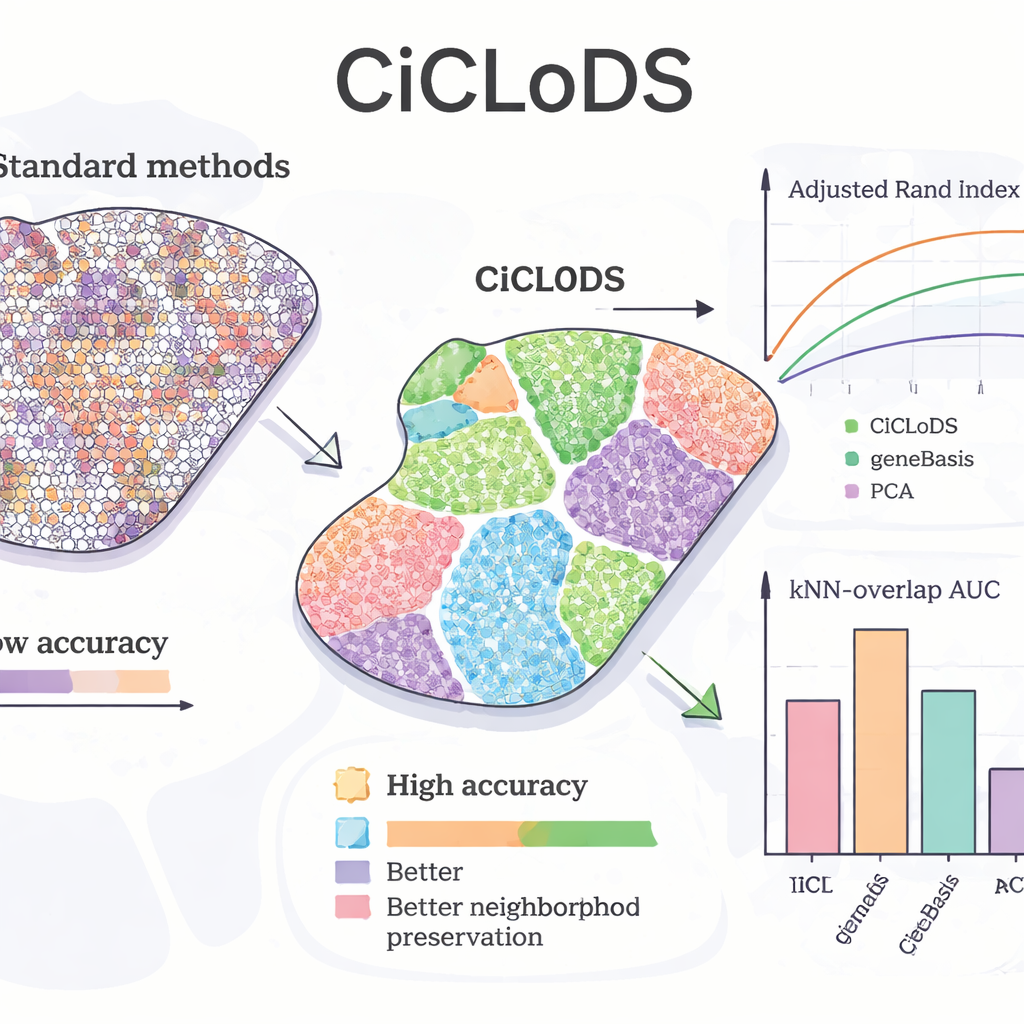
Generalizando entre cerebros y potenciando otros métodos
Para evaluar si CiCLoDS se mantiene en tejidos e individuos muy diferentes, los autores analizaron muestras de corteza prefrontal dorsolateral humana de tres donantes. Aquí, CiCLoDS se comportó a la par o mejor que métodos espaciales especializados como BayesCafe y BayesSpace, especialmente en una muestra difícil donde las otras herramientas tuvieron problemas. El estudio también destaca un uso “híbrido”: ejecutar CiCLoDS primero para obtener clústeres estables y después introducirlos en BayesSpace. Esta estrategia de inicio en caliente aumentó la precisión global y produjo patrones de capas cerebrales que mejor coincidieron con las anotaciones de expertos, mostrando que CiCLoDS puede funcionar de forma independiente y también hacer que modelos probabilísticos posteriores sean más fiables.
Por qué esto importa para la biología y la medicina
Para los no especialistas, la conclusión clave es que CiCLoDS convierte mapas celulares abrumadores en resúmenes concisos y biológicamente significativos. En lugar de trabajar con miles de mediciones ruidosas, los investigadores obtienen una lista manejable de genes y clústeres espaciales claros que reflejan la organización real del tejido: zonas metabólicas en el hígado, vasos sanguíneos y sus nichos, y estructuras en capas en el cerebro. Debido a que el presupuesto de genes lo controla el usuario y los cálculos son ligeros, CiCLoDS puede ayudar a diseñar paneles génicos dirigidos para experimentos futuros, guiar la interpretación de conjuntos de datos espaciales complejos y proporcionar puntos de partida robustos para modelados más elaborados. En una época en la que el cuello de botella ya no es la recolección de datos sino su comprensión, herramientas como CiCLoDS prometen hacer que los mapas tisulares de alta dimensionalidad sean tanto prácticos como reveladores.
Cita: Wang, N., He, Y., Ray, E. et al. CiCLoDS: Joint cell clustering and gene selection for single-cell spatial transcriptomics. Sci Rep 16, 5356 (2026). https://doi.org/10.1038/s41598-026-39168-1
Palabras clave: transcriptómica espacial, agrupación celular, selección de paneles de genes, arquitectura tisular, análisis unicelular