Clear Sky Science · es
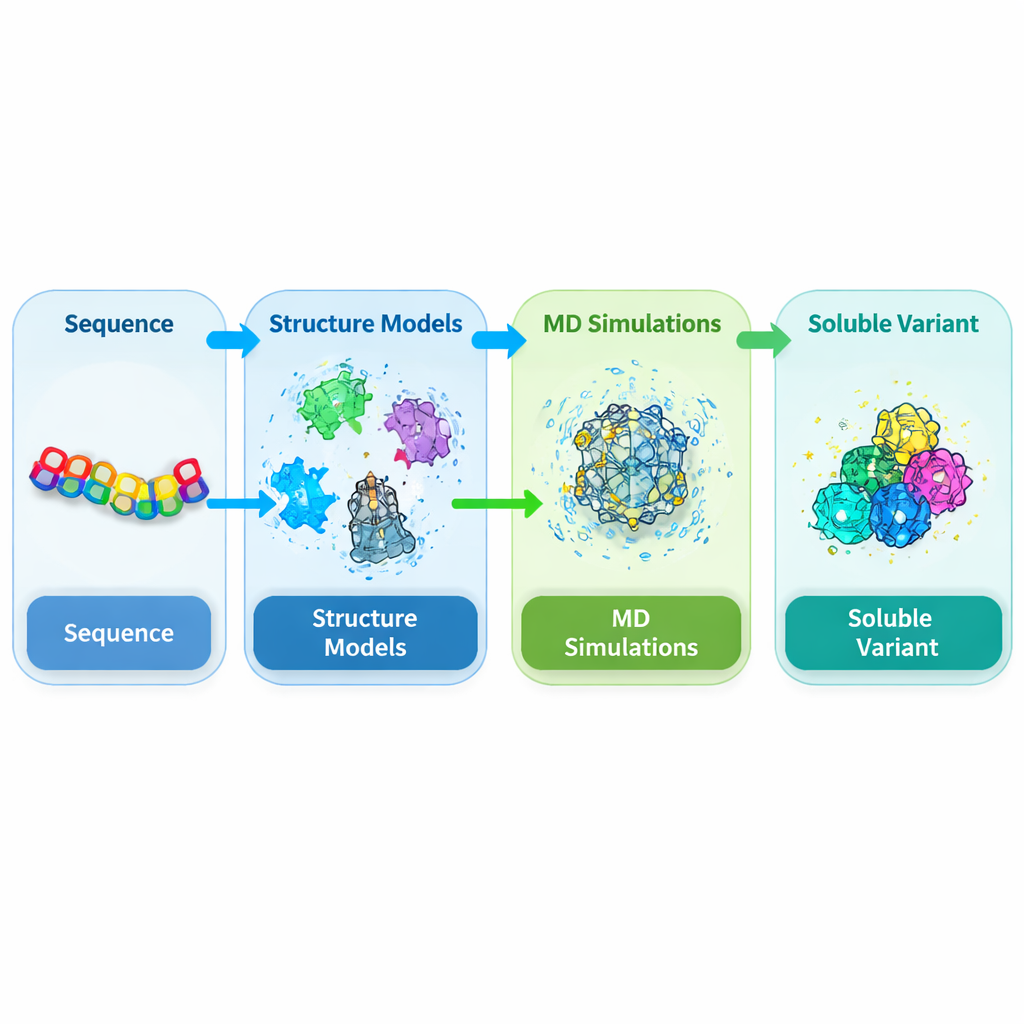
Optimización computacional de la solubilidad del dominio calpaína DEK1 mediante modelado estructural integrado y mutagénesis dirigida basada en datos
Por qué importa lograr que las proteínas vegetales se comporten
Muchas de las proteínas que controlan el crecimiento de las plantas son moléculas grandes y frágiles que se niegan a disolverse cuando los científicos intentan estudiarlas en el laboratorio. Una de estas proteínas, llamada DEK1, ayuda a dar forma al cuerpo de la planta desde el nivel de las células individuales hacia arriba. Pero debido a que una parte crucial de DEK1 se agregaba cuando se producía en bacterias, su estructura tridimensional ha permanecido desconocida, lo que ralentiza los esfuerzos por comprenderla y aprovecharla. Este estudio muestra cómo el modelado por ordenador y el diseño inteligente basado en datos pueden rediseñar esa región problemática para que sea más soluble, sin romper su arquitectura—ofreciendo una receta general para domesticar proteínas difíciles.
Apuntar al punto problemático en una proteína vegetal clave
DEK1 es una proteína inusualmente grande incrustada en las membranas celulares y rematada por una región enzimática de corte conocida como dominio calpaína. Trabajos genéticos han demostrado que este dominio es esencial para el desarrollo normal en plantas como los musgos y los cultivos, sin embargo su estructura nunca se ha resuelto experimentalmente. Cuando los investigadores intentan producir este núcleo calpaína (llamado CysPc) en la bacteria hospedadora común Escherichia coli, tiende a volverse insoluble y forma densos cuerpos de inclusión. Eso hace casi imposible purificarlo en las cantidades y la calidad necesarias para estudios estructurales y funcionales detallados. Por ello, los autores se propusieron rediseñar el dominio CysPc para que se disolviera más fácilmente conservando su forma global.
Construir un modelo 3D fiable desde cero
Dado que no existe una estructura experimental para esta calpaína vegetal, el equipo tuvo que predecir primero su forma tridimensional. Combinó varias herramientas de predicción de estructura de última generación, incluidas AlphaFold2, SWISS-MODEL e I-TASSER, y ancló esas predicciones a estructuras conocidas de calpaínas mamíferas relacionadas. Usando un enfoque de consenso, refinaron y comprobaron los modelos resultantes con múltiples pruebas de calidad que evalúan la geometría de la cadena principal, el empaquetamiento y la concordancia con patrones estructurales conocidos. Estas comprobaciones independientes mostraron que el modelo integrado del dominio CysPc era más fiable que cualquier predicción individual, proporcionando un punto de partida sólido para explorar cómo pequeños cambios en la secuencia de aminoácidos podrían mejorar la solubilidad.
Probar mutaciones virtuales en un solvente simulado
Con el modelo 3D en mano, los autores realizaron extensas simulaciones de dinámica molecular, en las que la proteína y las moléculas de agua circundantes se siguen en el tiempo en el ordenador. Se centraron en residuos de la superficie proteica que eran flexibles, hidrófobos o se predecía que promovían la agregación. Las posiciones candidatas se mutaron individualmente por aminoácidos más compatibles con el agua y luego se simularon durante 200 nanosegundos cada una. Para cada variante midieron características relacionadas con la solubilidad, como cuánta superficie está en contacto con el agua, cuán compacta permanece la proteína y con qué intensidad fluctúan los átomos. Muchas mutaciones simples aumentaron modestamente la exposición al solvente o el número de enlaces de hidrógeno internos sin alterar el pliegue general, lo que sugiere que el armazón básico de CysPc podría tolerar sustituciones elegidas con cuidado.
Dejar que los algoritmos exploren el espacio de mutaciones
Cambiar solo un residuo rara vez produce mejoras drásticas en la solubilidad, por lo que los investigadores exploraron a continuación combinaciones de dos y tres mutaciones. Generaron una biblioteca de variantes dobles y triples a partir de las mejores mutaciones simples y de nuevo simularon cada una. Para puntuar y clasificar estos diseños de forma justa, definieron un índice ponderado que combina múltiples características de las simulaciones conocidas por correlacionar con la solubilidad, premiando el aumento de hidratación y enlaces internos y penalizando la flexibilidad excesiva. Luego utilizaron un algoritmo de aprendizaje por refuerzo (Proximal Policy Optimization) para navegar el enorme espacio de posibles mutantes triples y proponer las combinaciones más prometedoras. Esta búsqueda basada en datos convergió en un mutante triple particular, llamado MUT347, como el candidato principal.
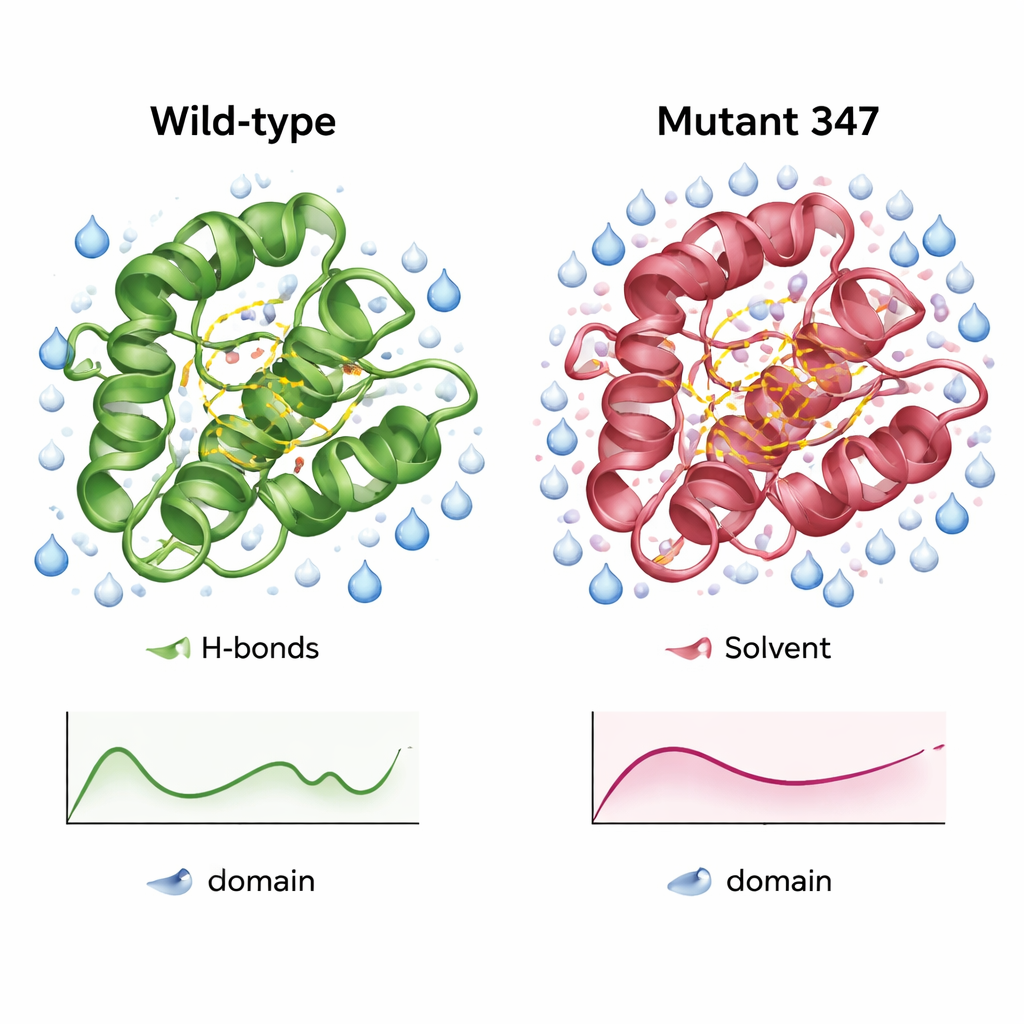
Una versión más compacta y mejor hidratada de la enzima
Simulaciones detalladas del dominio CysPc de tipo salvaje y de MUT347 revelaron en qué difería la variante diseñada. MUT347 se equilibró más rápidamente y mostró desviaciones totales menores respecto a su forma inicial, lo que indica mayor estabilidad estructural en solución. Sus bucles y extremos de cadena fueron algo menos flexibles, mientras que la región catalítica central conservó su flexibilidad original, lo que sugiere que los movimientos funcionalmente importantes se preservaron. El mutante triple presentó más enlaces de hidrógeno internos y una mayor superficie accesible al agua en regiones clave, señales de una superficie mejor organizada y más hidratada. Bajo distintas concentraciones de sal y niveles de pH, MUT347 mantuvo consistentemente fluctuaciones más bajas que la proteína original, un comportamiento asociado con una menor tendencia a agregarse.
Qué significa esto para estudiar y reutilizar proteínas
Para el público no especializado, la conclusión es que los autores han construido una receta mayormente computacional para convertir un fragmento torpe y propenso a agregarse de una proteína vegetal vital en una versión más soluble y manejable, sin tener que conocer su estructura experimentalmente de antemano. Al combinar predicción de estructuras moderna, simulaciones de larga escala temporal y algoritmos de aprendizaje capaces de gestionar muchas decisiones de diseño a la vez, identificaron una triple mutación que se predice estabiliza el pliegue y lo expone de manera más favorable al agua. Aunque aún se necesitan trabajos experimentales para confirmar las mejoras en tubos de ensayo reales, este marco podría ser de utilidad general para rescatar otras proteínas eucariotas difíciles de producir, ayudando en última instancia a los científicos a desbloquear estructuras y funciones que hoy están fuera de alcance.
Cita: Dabiri, M., Levarski, Z., Struhárňanská, E. et al. Computational optimization of DEK1 calpain domain solubility through integrated structural modelling and data-driven targeted mutagenesis. Sci Rep 16, 7767 (2026). https://doi.org/10.1038/s41598-026-38805-z
Palabras clave: solubilidad de proteínas, mutagénesis computacional, dinámica molecular, calpaína DEK1 de plantas, ingeniería de proteínas