Clear Sky Science · es
El lncRNA FTX promueve la fibrosis miocárdica al actuar como esponja de miR-335-3p para regular la señalización TFEC/ILK
Por qué importa la cicatrización del corazón
La insuficiencia cardiaca afecta a decenas de millones de personas en todo el mundo y con frecuencia se desarrolla de forma silenciosa durante años. Un motor importante de este deterioro es la fibrosis miocárdica: la formación lenta y progresiva de tejido cicatricial en el músculo cardiaco, que lo vuelve más rígido y menos capaz de bombear sangre. Este estudio explora el «cableado» molecular que indica a las células cardiacas que depositen tejido cicatricial en exceso, e identifica una nueva cadena de moléculas que podría apuntarse para frenar o incluso revertir este proceso dañino.
Una mirada más cercana a la cicatrización cardiaca
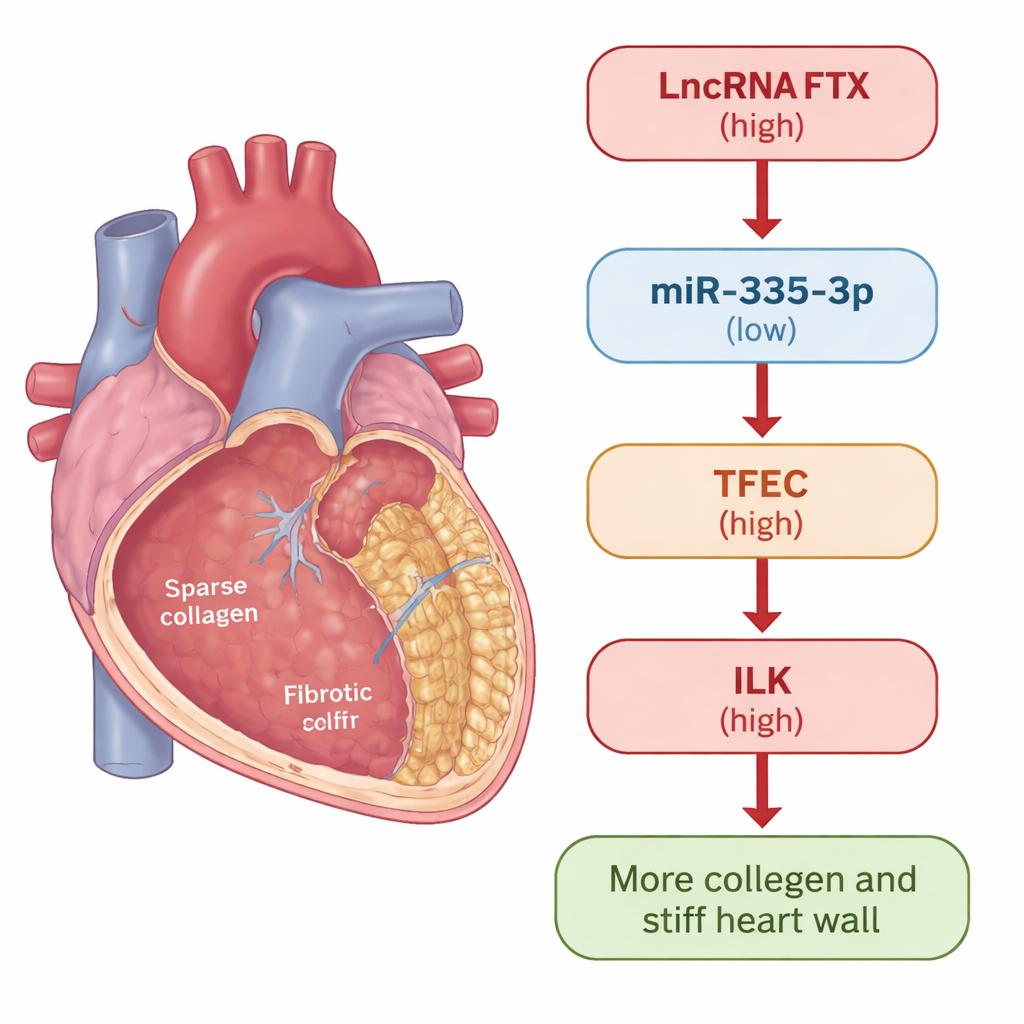
Cuando el corazón se lesiona o se somete a estrés, células de apoyo llamadas fibroblastos cardíacos entran en acción. En una reparación saludable ayudan a parchear el daño. Pero en enfermedad crónica pueden cambiar a un estado sobreactivado, produciendo colágeno en exceso y otros componentes de la matriz extracelular, lo que acaba rigidizando la pared cardiaca. Los investigadores usaron dos modelos para estudiar este proceso: ratones tratados con isoproterenol, que induce de forma fiable la fibrosis cardiaca, y fibroblastos cardíacos humanos expuestos a la molécula de señalización TGF-β1, un desencadenante conocido de la cicatrización. En ambos escenarios midieron cómo cambiaban genes y proteínas específicos a medida que se desarrollaba la fibrosis.
Una reacción en cadena dañina dentro de las células
El equipo se centró en un factor de transcripción llamado TFEC, una proteína que reside en el núcleo celular y activa otros genes. Encontraron que TFEC, junto con otra proteína llamada quinasa ligada a integrinas (ILK), aumentaba de forma sostenida cuando los fibroblastos se orientaban hacia un estado fibrótico formador de cicatrices. Silenciar TFEC o ILK redujo drásticamente marcadores clásicos de fibrosis como la actina α de músculo liso y los colágenos I y III, y también atenuó una vía de control del crecimiento (Akt/GSK3β y la señalización Hippo) conocida por promover la cicatrización tisular. Experimentos de mapeo de unión al ADN mostraron que TFEC se une directamente al promotor del gen ILK y potencia su actividad, situando a TFEC claramente río arriba de ILK en una cascada de señalización pro‑fibrosis.
Interruptores de ARN que controlan al regulador maestro
Para entender qué controla al propio TFEC, los investigadores recurrieron a ARNs no codificantes: moléculas de ARN que no forman proteínas pero actúan como afinadores finos de la actividad génica. Identificaron un ARN pequeño, miR‑335‑3p, que estaba reducido en corazones y células fibróticas. Elevar los niveles de miR‑335‑3p disminuía TFEC, mientras que bloquearlo lo aumentaba, y ensayos reporteros confirmaron que miR‑335‑3p se une directamente a los mensajes de TFEC para mantenerlos bajo control. A continuación hallaron un ARN largo no codificante, llamado FTX, que estaba elevado en la fibrosis y que interactuaba físicamente con miR‑335‑3p. FTX actuaba como una esponja molecular: absorbía miR‑335‑3p, impidiendo que este ARN pequeño frenara a TFEC. Como resultado, TFEC e ILK aumentaban y los fibroblastos producían más colágeno formador de cicatrices.
Del cultivo celular a corazones vivos
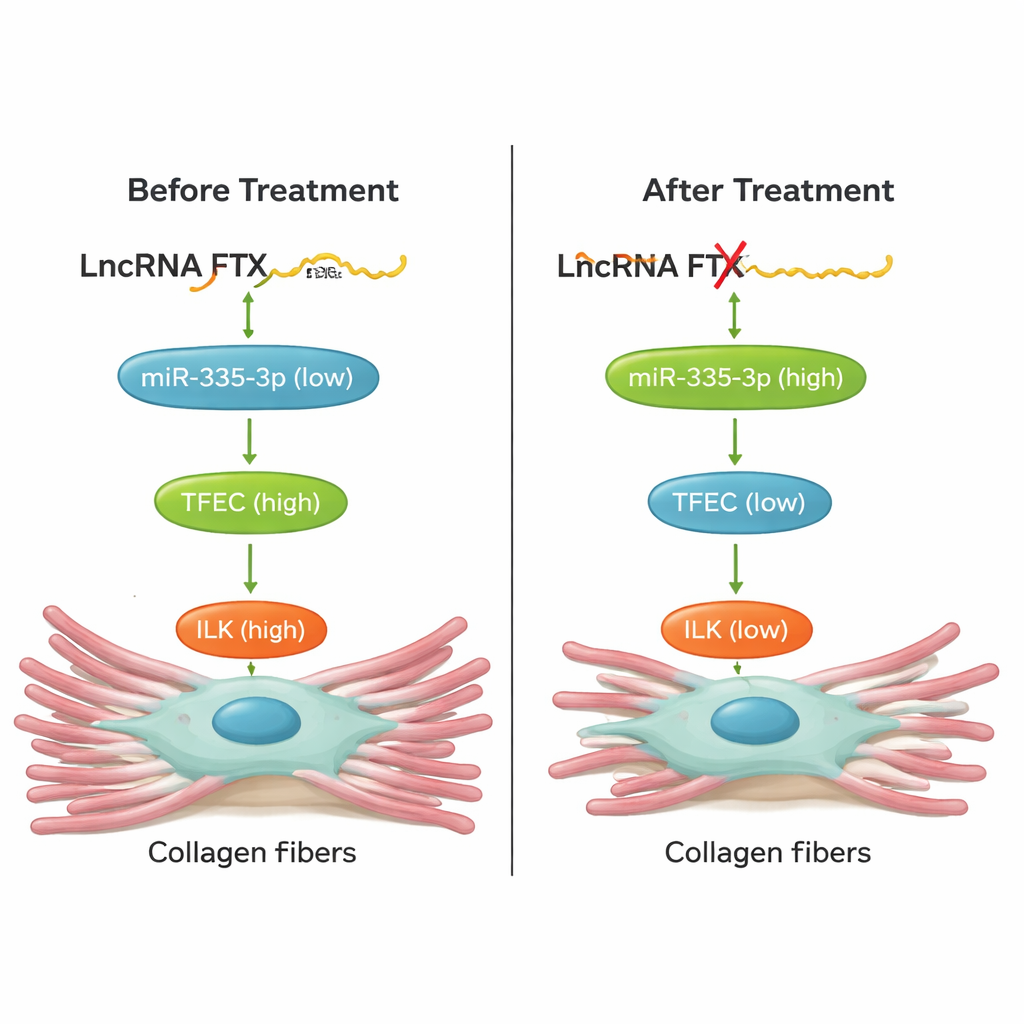
De forma crucial, el equipo probó si interrumpir esta cadena podía proteger realmente corazones en animales. En ratones expuestos a isoproterenol, reducir TFEC, disminuir FTX en el corazón mediante un vector de terapia génica AAV9, o aumentar miR‑335‑3p con un «agomir» químicamente estabilizado condujo a menor acumulación de colágeno y a niveles más bajos de marcadores de fibrosis en el tejido cardiaco. Estas intervenciones también mejoraron la función cardiaca: el volumen sistólico y la fracción de eyección regresaron hacia valores normales, y se atenuaron incrementos nocivos de la frecuencia cardiaca. Experimentos de rescate en células mostraron que alterar un componente del eje FTX/miR‑335‑3p/TFEC/ILK desplazaba de manera predecible a los demás, confirmando que se trata de una vía estrechamente conectada y no de una correlación casual.
Qué significa esto para tratamientos futuros
Para un público no especializado, la conclusión es que los autores han identificado una nueva «palanca de control» de la cicatrización cardiaca. Un ARN largo llamado FTX levanta el freno (miR‑335‑3p) sobre un interruptor maestro (TFEC), que a su vez activa ILK y señales pro‑cicatrización aguas abajo, impulsando la deposición excesiva de colágeno y la rigidez del corazón. Al reducir FTX, restaurar miR‑335‑3p o bloquear directamente TFEC, fue posible en ratones disminuir la fibrosis y mejorar la función de bombeo. Aunque se necesita más trabajo para confirmar esta vía en pacientes humanos y desarrollar terapias seguras, esta cadena reguladora basada en ARN ofrece varios puntos prometedores para intervenir en la insuficiencia cardíaca impulsada por la fibrosis.
Cita: Yao, F., He, Z., Zheng, C. et al. LncRNA FTX promotes myocardial fibrosis by sponging miR-335-3p to regulate TFEC/ILK signaling. Sci Rep 16, 7340 (2026). https://doi.org/10.1038/s41598-026-38615-3
Palabras clave: fibrosis miocárdica, insuficiencia cardíaca, ARN no codificante, fibroblastos cardíacos, señalización de la fibrosis