Clear Sky Science · es
Potencial interatómico por aprendizaje automático para las propiedades estructurales de los óxidos de hierro
Por qué importan las rocas oxidadas
Los óxidos de hierro —los minerales que dan el color al óxido— sostienen discretamente gran parte de la vida moderna. Son la principal fuente de hierro para el acero, ingredientes clave en baterías y células solares, e incluso ayudan a depurar agua contaminada. Sin embargo, a pesar de su importancia, todavía nos cuesta predecir cómo se comportan estos materiales en condiciones reales, especialmente a escala atómica. Este artículo describe cómo investigadores emplearon inteligencia artificial moderna para construir un modelo digital rápido y preciso de un óxido de hierro crucial, la hematita, abriendo la puerta a experimentos virtuales más fiables en todo, desde el procesamiento de mineral hasta dispositivos de energía limpia.
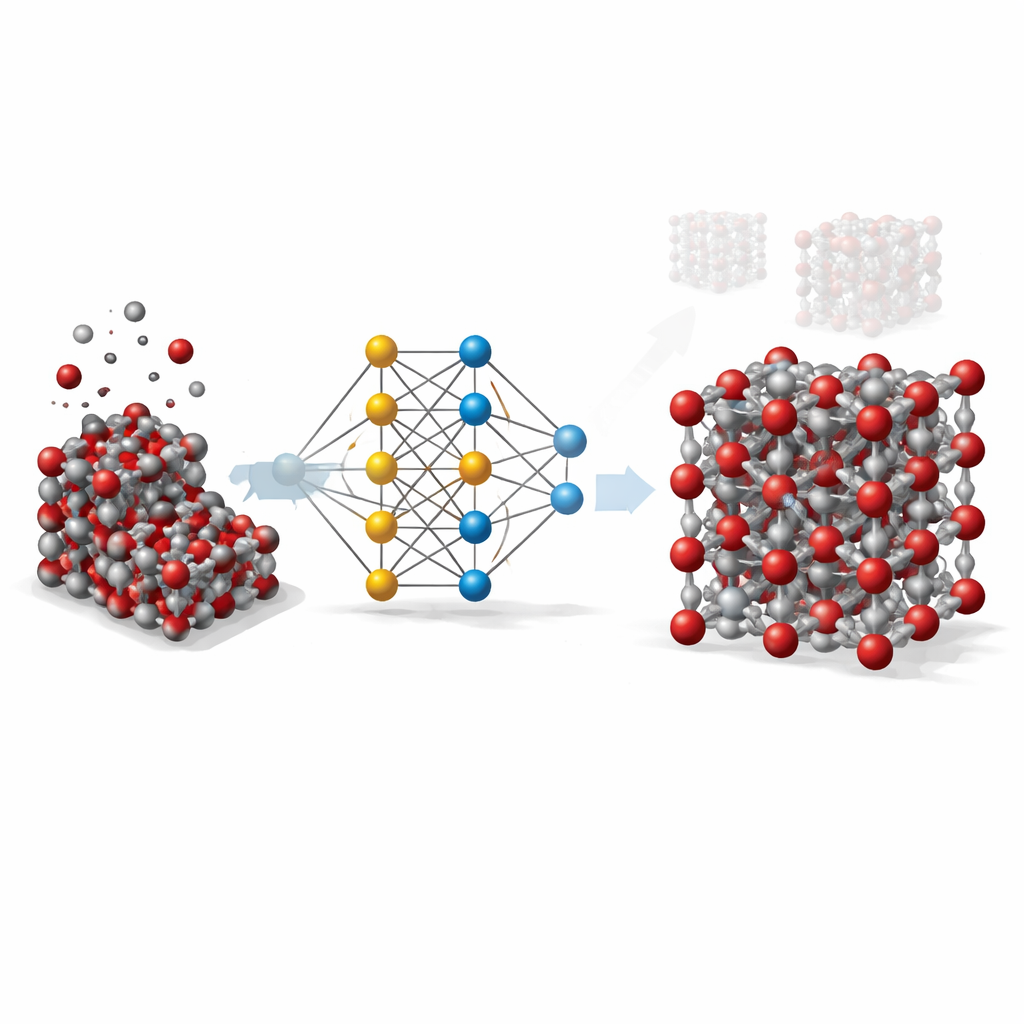
De cálculos costosos a atajos inteligentes
Para comprender un sólido como la hematita en detalle, los científicos idealmente recurren a métodos cuántico-mecánicos que siguen cómo interactúan electrones y átomos. Estos métodos, aunque muy precisos, son tan costosos computacionalmente que resultan imprácticos para simular muestras grandes o tiempos largos. Los modelos clásicos, en cambio, son rápidos pero toscos: se basan en fórmulas simples ajustadas a situaciones específicas y con frecuencia fallan cuando cambian temperatura, presión o la forma del cristal. El trabajo aquí presentado busca cerrar esa brecha usando aprendizaje automático para imitar la precisión de los cálculos cuánticos manteniendo la velocidad de los modelos tradicionales.
Enseñar a una red neuronal sobre los átomos
El equipo construyó lo que se conoce como un potencial de red neuronal de grafos para la hematita. En este enfoque, cada átomo se trata como un nodo en una red, y los enlaces y átomos vecinos son las conexiones entre nodos. Para enseñar a esta red cómo se atraen y repelen los átomos en la hematita, los investigadores generaron primero miles de instantáneas atómicas usando simulaciones estándar a lo largo de una amplia gama de temperaturas, presiones y distorsiones cristalinas, incluyendo tanto cristales en volumen como superficies expuestas. Luego usaron un método cuántico avanzado (DFT+U) para calcular la energía, las fuerzas y las tensiones internas de cada instantánea, y entrenaron la red neuronal para reproducir esos valores lo más fielmente posible.
Comprobar el modelo frente a la realidad
Una vez entrenado, el nuevo potencial —denominado Fe-MLIP— fue sometido a pruebas rigurosas. Los autores compararon sus predicciones para magnitudes estructurales básicas, como las dimensiones de la red y cómo se deforma el cristal bajo esfuerzo, con experimentos y con varios modelos clásicos de uso extendido. Fe-MLIP reprodujo la conocida estructura cristalina de la hematita con un error de solo unos pocos porcentajes y capturó su comportamiento elástico casi tan bien como los cálculos cuánticos directos, superando claramente a otros campos de fuerza para muchas propiedades. También obtuvo buenos resultados en pruebas más sutiles, como la expansión térmica y las vibraciones atómicas, importantes para el transporte de calor y la espectroscopía. Estas frecuencias vibracionales, que nunca se mostraron explícitamente durante el entrenamiento, se acercaron más a los valores medidos que las predichas por modelos competidores.
Ir más allá de un solo mineral
Los investigadores exploraron después hasta qué punto se podía aplicar este modelo basado en hematita. Lo usaron en óxidos de hierro relacionados —maghemita y magnetita— que comparten bloques atómicos similares pero difieren en la disposición cristalina y en los estados de carga del hierro. Aunque Fe-MLIP no fue entrenado con estas fases, produjo valores razonables para sus tamaños de celda y rigideces, a menudo igualando o superando a modelos clásicos especializados. El potencial también capturó la estabilidad relativa de superficies cristalinas clave e incluso las tendencias en el costo energético de crear vacantes atómicas, rasgos cruciales para entender la corrosión, la catálisis y el rendimiento en baterías.
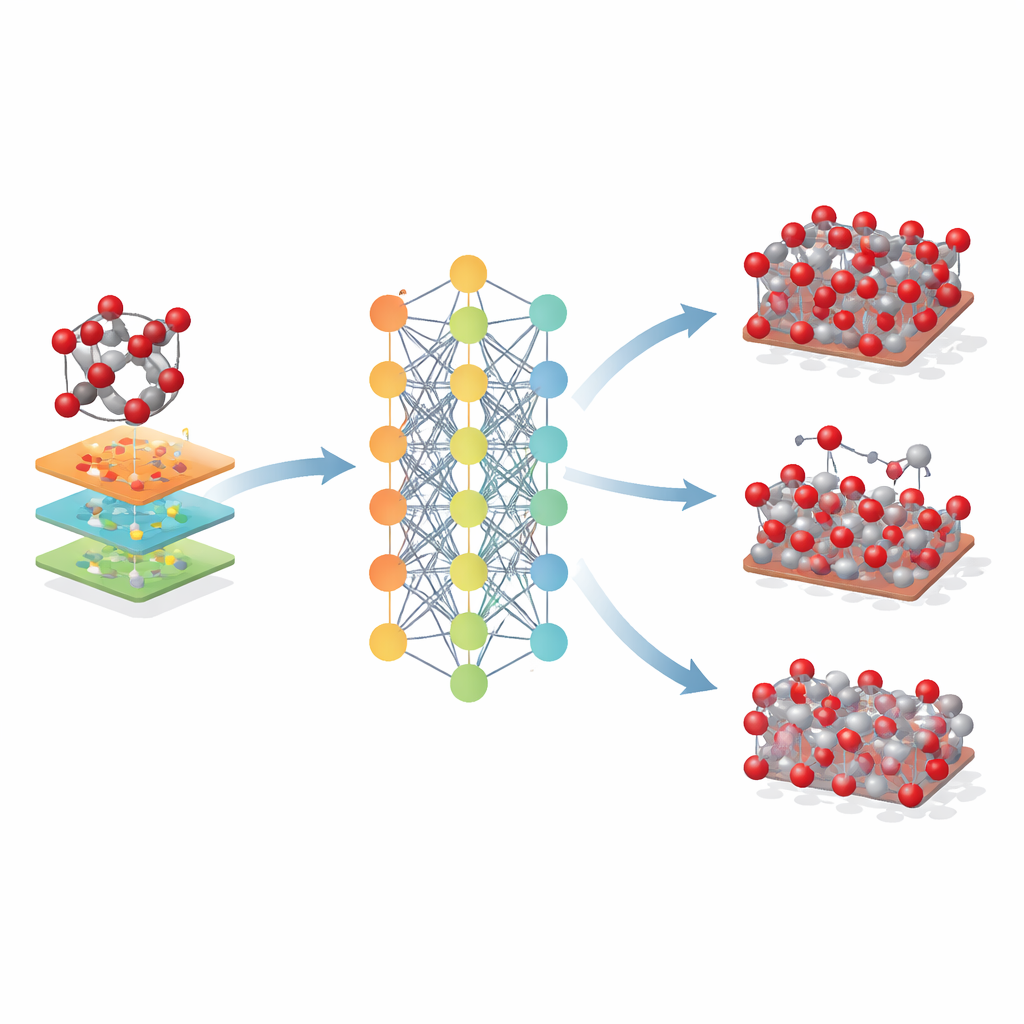
Qué implica esto para el diseño futuro de materiales
Para el público general, la conclusión es que este trabajo proporciona un potente «gemelo digital» para los óxidos de hierro. El modelo Fe-MLIP permite a los investigadores ejecutar simulaciones grandes y de larga duración de la hematita y materiales relacionados con una fiabilidad próxima al nivel cuántico pero a una fracción del coste. Si bien hereda algunas limitaciones del método cuántico subyacente y actualmente está centrado en hierro y oxígeno, ya permite estudios más realistas de cómo estos minerales responden a esfuerzos, calor, superficies y defectos. En términos prácticos, una herramienta así puede acelerar el diseño de procesos siderúrgicos mejores, catalizadores y baterías más eficientes, y tecnologías ambientales mejoradas que dependen de los óxidos de hierro —todo ello permitiendo a los científicos probar ideas en el ordenador antes de acudir al laboratorio o a la mina.
Cita: Torres, A., de Oliveira, A.B., Barbosa, M.d.S. et al. Machine learning interatomic potential for the structural properties of iron oxides. Sci Rep 16, 8576 (2026). https://doi.org/10.1038/s41598-026-38096-4
Palabras clave: hematita, óxidos de hierro, potencial de aprendizaje automático, redes neuronales de grafos, dynamics molecular