Clear Sky Science · es
Caracterización molecular de trastornos atáxicos y neuropáticos de herencia recesiva en familias paquistaníes consanguíneas
Por qué esto importa para las familias y los médicos
Los problemas con el equilibrio, la marcha y la sensibilidad en manos y pies pueden ser profundamente discapacitantes, sobre todo cuando comienzan en la infancia y empeoran de forma lenta a lo largo de la vida. Para muchas familias, especialmente en regiones donde los matrimonios entre primos son frecuentes, estos síntomas se repiten durante varias generaciones sin una explicación clara. Este estudio aborda una cuestión urgente para esas familias en Pakistán: ¿puede el análisis moderno del ADN revelar por fin las causas genéticas ocultas de su ataxia (pobre equilibrio y coordinación) y neuropatía periférica (daño a los nervios de las extremidades), y ayudar a los médicos a ofrecer diagnósticos más precisos y posibles tratamientos?
Rastreando la enfermedad hereditaria en familias numerosas
Los investigadores trabajaron con siete familias paquistaníes extendidas en las que varios miembros presentaban problemas graves de movimiento y nervios. Algunas personas tenían principalmente ataxia, lo que dificultaba caminar con estabilidad o controlar el habla y los movimientos oculares. Otras mostraban signos clásicos de neuropatía periférica, como pérdida de masa muscular en manos y pies, deformidades del pie y pérdida de reflejos. En estas familias, los padres estaban emparentados entre sí, lo que aumentaba la probabilidad de que los hijos heredaran dos copias del mismo gen raro defectuoso. Usando muestras de sangre de parientes afectados y no afectados, el equipo realizó secuenciación del exoma—leyendo casi todas las partes codificantes de proteínas del genoma—para buscar cambios dañinos que siguieran el patrón de la enfermedad a través de los árboles genealógicos. 
Localizando genes raros defectuosos
Al filtrar las diferencias de ADN comunes y benignas, los científicos hallaron variantes probablemente causantes de la enfermedad en cinco de las siete familias. Cada una de estas familias portaba su propio cambio genético específico, y todas seguían un patrón recesivo: la enfermedad se manifestaba solo cuando se heredaban dos copias defectuosas, una de cada progenitor. En una familia con problemas de equilibrio de inicio en la edad adulta y dificultades del habla, el responsable fue una variante rara en un gen llamado MFSD8, que ayuda a transportar materiales hacia compartimentos de reciclaje celular llamados lisosomas. En otra familia, un cambio dañino en AFG3L2, una proteína que mantiene la salud de las mitocondrias—las centrales energéticas de la célula—se asoció con ataxia espástica de inicio infantil con distonía (contracciones musculares anormales). Una tercera familia portaba un error de cambio de marco (frameshift) en SETX, un gen que protege el ADN durante la reparación y que ya se conoce por causar ataxia con apraxia oculomotora, un trastorno que también afecta los movimientos oculares.
Una mirada más cercana al daño nervioso hereditario
Dos familias adicionales presentaban una forma de la enfermedad de Charcot‑Marie‑Tooth (CMT), un grupo de trastornos hereditarios que dañan los nervios largos hacia los pies y las manos. En ambas, los investigadores encontraron variantes dañinas en un gen llamado GDAP1, que es crucial para la función mitocondrial normal en las células nerviosas. Un cambio en GDAP1 acortaba la proteína y se asoció con una enfermedad muy grave y de inicio temprano; otro sustituyó un solo bloque constructivo de la proteína y produjo una evolución algo más leve. De forma notable, el paciente más gravemente afectado en una familia con CMT era también homocigoto para una variante conocida causante de enfermedad en un segundo gen, MMACHC, que participa en el procesamiento de la vitamina B12 y que en algunos casos puede tratarse con terapias basadas en vitaminas. Este doble impacto podría explicar por qué sus síntomas fueron peores que los de sus parientes que no tenían la variante en MMACHC.
Cuando la búsqueda del ADN no basta
No todas las familias ofrecieron una respuesta genética clara. En dos de las siete familias, el equipo no pudo encontrar ningún cambio único en el exoma que coincidiera de forma convincente con el patrón de enfermedad. En un caso, identificaron una variante en el gen EHHADH que seguía el patrón de herencia pero se predice como inocua y se sabe que causa una condición renal diferente cuando está alterado. En otro, dos primos con problemas de movimiento similares resultaron tener causas subyacentes distintas: un niño portaba una variante dañina conocida en ALS2, que puede conducir a formas juveniles de enfermedad de la neurona motora, mientras que sus primos afectados no la tenían. Estos casos no resueltos sugieren que mutaciones importantes pueden hallarse en regiones del genoma que la secuenciación del exoma estándar no captura, o que más de un factor genético sutil podría estar interactuando.
Qué significa esto para los pacientes y la atención futura
En conjunto, los resultados muestran que las herramientas genéticas potentes pueden descubrir los genes específicos detrás de trastornos complejos de nervios y equilibrio, incluso en entornos con recursos limitados. Para las cinco familias con hallazgos claros, el trabajo transforma etiquetas vagas como «ataxia» o «neuropatía» en diagnósticos precisos ligados a genes concretos, lo que puede orientar el consejo genético, informar sobre el pronóstico y, en algunos casos, destacar opciones de tratamiento, como las terapias relacionadas con la vitamina B12 para la enfermedad ligada a MMACHC. El estudio también amplía la comprensión científica de cómo genes como MFSD8, AFG3L2, SETX, GDAP1, MMACHC y ALS2 condicionan la salud de las células nerviosas en el cerebro, la médula espinal y los nervios periféricos. De cara al futuro, será necesaria una secuenciación genómica más completa y estudios funcionales para resolver los misterios pendientes y traducir estos hallazgos genéticos en un diagnóstico más precoz y una mejor atención para los niños y adultos afectados. 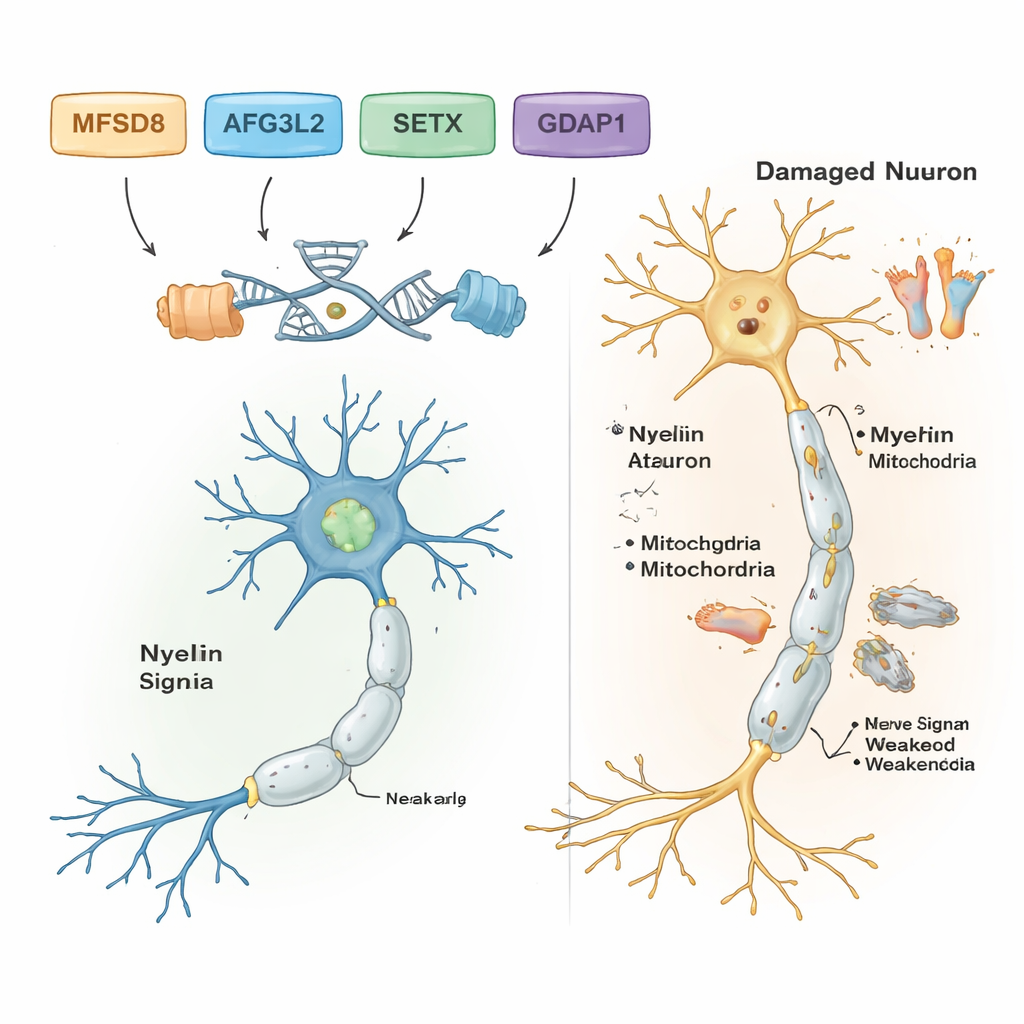
Cita: Aslam, F., Wajid, M., Butt, A.I. et al. Molecular characterization of recessively inherited ataxic and neuropathic disorders in consanguineous Pakistani families. Sci Rep 16, 6529 (2026). https://doi.org/10.1038/s41598-026-37808-0
Palabras clave: ataxia, neuropatía periférica, secuenciación del exoma, enfermedad de Charcot‑Marie‑Tooth, diagnóstico genético