Clear Sky Science · es
Optimización de la selección de características en datos de microarrays de cáncer mediante un marco evolutivo impulsado por montículos para espacios de alta dimensionalidad
Por qué importa elegir los genes correctos
Las pruebas de cáncer basadas en tecnologías genéticas modernas pueden medir decenas de miles de genes a la vez, pero los médicos suelen disponer de datos de solo unas pocas docenas de pacientes. Ocultas en esta enorme “selva génica” hay señales mucho más reducidas que realmente distinguen un tipo de cáncer de otro, o un tumor de tejido sano. Este artículo presenta un nuevo método de búsqueda inteligente para seleccionar automáticamente esos genes clave, con el objetivo de hacer que el diagnóstico asistido por ordenador sea más preciso, rápido y fácil de interpretar.
Demasiadas señales, muy pocos datos
Los experimentos con microarrays y tecnologías similares permiten medir los niveles de actividad de miles de genes en cada muestra de paciente. Sin embargo, el número de muestras suele ser muy pequeño, a veces menos de cien. Muchas de estas mediciones génicas son ruidosas, redundantes o irrelevantes para la enfermedad en cuestión. Conservarlas todas puede abrumar a los algoritmos de aprendizaje, ralentizar los cálculos y producir modelos engañosos que se agarran a peculiaridades aleatorias en lugar de a la biología verdadera. El proceso de reducir esto a un subconjunto útil se llama “selección de características”, y es crucial si queremos predicciones fiables a partir de datos médicos de alta dimensionalidad.
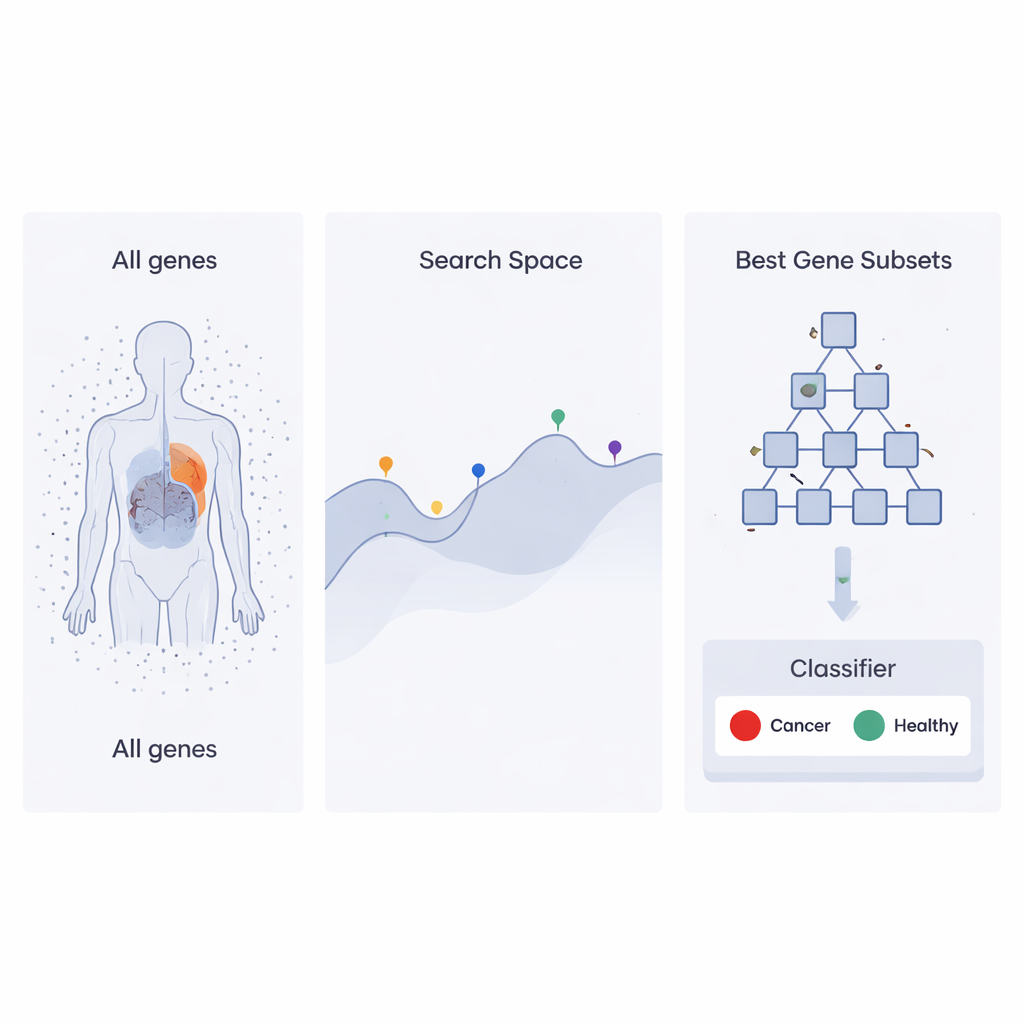
Una estrategia de búsqueda inspirada en las jerarquías empresariales
Los autores se basan en un enfoque de optimización reciente llamado Heap‑Based Optimizer (HBO), que toma ideas de cómo se organiza el personal en una empresa. Imagine cada posible conjunto de genes como un “empleado” cuyo desempeño se juzga por cuánto ayuda a un clasificador a distinguir muestras cancerosas de sanas. Estos empleados se disponen en una jerarquía, como una escalera corporativa, usando una estructura informática conocida como montículo (heap). Los conjuntos de genes de alto rendimiento están cerca de la cima, mientras que los más débiles quedan más abajo. A lo largo de muchas rondas, los empleados peor posicionados ajustan sus elecciones copiando y modificando ligeramente lo que hacen sus jefes y colegas, empujando gradualmente a toda la organización hacia mejores soluciones.
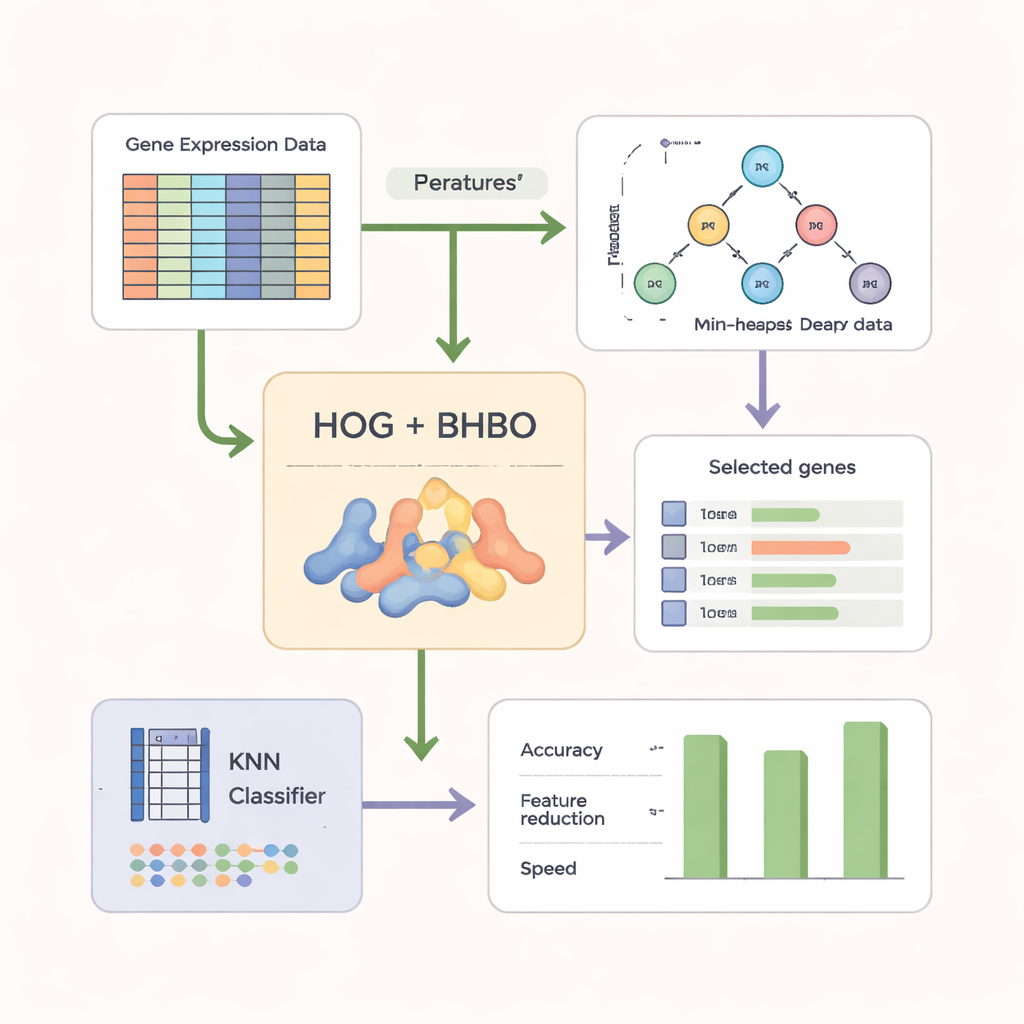
Convertir datos génicos crudos en patrones más nítidos
Para hacer la búsqueda más eficaz, los autores no se basan solo en las lecturas génicas crudas. Primero remodelan los datos de microarrays en una forma similar a una imagen y aplican una técnica llamada Histograma de Gradientes Orientados (HOG), ampliamente usada en visión por ordenador. HOG captura cómo cambian los niveles de expresión a lo largo de los genes, resaltando patrones locales en lugar de mediciones aisladas. Estas características basadas en patrones se combinan luego con la información génica original. Un clasificador simple llamado k‑Nearest Neighbors (KNN) actúa como el “juez”, puntuando cada subconjunto candidato de genes por la precisión con la que etiqueta nuevas muestras y también recompensando conjuntos más pequeños y compactos.
Pruebas en múltiples conjuntos de datos de cáncer
Los investigadores evaluaron su versión binaria del Heap‑Based Optimizer (BHBO) en nueve conjuntos de datos públicos de microarrays de cáncer, incluidos tumores cerebrales, leucemias, cáncer de próstata y colecciones mixtas con muchos subtipos. Cada conjunto tenía desde miles hasta más de quince mil genes medidos, pero relativamente pocas muestras de pacientes. Para cada conjunto, BHBO se ejecutó repetidamente y se comparó con siete métodos de búsqueda bien conocidos, como algoritmos genéticos y optimización por enjambre de partículas. El equipo midió no solo la precisión, sino también cuántos genes se mantenían, la rapidez con la que convergía la búsqueda y la estabilidad de los resultados cuando los datos se perturbaban con ruido simulado, efectos de lote y errores de etiquetado.
Lo que logró el nuevo método
En los nueve conjuntos de datos, el enfoque impulsado por montículos alcanzó una precisión media de clasificación de aproximadamente el 95 por ciento mientras reducía el número de genes en más del 85 por ciento. Superó claramente a métodos competidores en varios conjuntos y mostró una convergencia más rápida, lo que significa que encontró buenos conjuntos de genes en menos pasos de búsqueda. Incluso cuando los autores corrompieron deliberadamente los datos —añadiendo ruido o invirtiendo algunas etiquetas de muestra— el rendimiento del método cayó solo ligeramente y se mantuvo por encima de las alternativas. Las pruebas estadísticas confirmaron que estas mejoras difícilmente se debían al azar.
Qué significa esto para el futuro del diagnóstico del cáncer
En términos prácticos, este trabajo demuestra que una estrategia de búsqueda bien diseñada puede cribar enormes conjuntos de datos genéticos y descubrir paneles pequeños y ricos en información de genes que todavía clasifican bien los cánceres. Para clínicos e investigadores, conjuntos génicos compactos son más fáciles de validar biológicamente, más baratos de medir en pruebas complementarias y más adecuados para integrarse en herramientas de apoyo a la decisión. Si bien el método no descubre directamente nuevos fármacos o vías, enfoca con mayor precisión los marcadores genéticos prometedores, ayudando a que otros estudios se centren en las señales más informativas ocultas en datos de cáncer de alta dimensionalidad.
Cita: Alweshah, M., Jebril, H., Kassaymeh, S. et al. Optimizing feature selection in cancer microarray data using a heap-driven evolutionary framework for high-dimensional spaces. Sci Rep 16, 6726 (2026). https://doi.org/10.1038/s41598-026-37803-5
Palabras clave: microarray de cáncer, selección de características, optimización metaheurística, biomarcadores génicos, minería de datos médicos