Clear Sky Science · es
Un método para la detección de variantes estructurales usando la matriz de contactos Hi-C y redes neuronales
Por qué importa que el ADN se doble en 3D
Nuestro ADN suele representarse como una simple cadena de letras, pero dentro de cada célula se pliega en una forma tridimensional compleja. Cuando grandes fragmentos de esta cadena se eliminan, se invierten o se reubican —cambios llamados variantes estructurales— pueden alterar genes y contribuir al desarrollo del cáncer. Este estudio presenta VarHiCNet, un nuevo sistema de inteligencia artificial que lee mapas de plegamiento del ADN en 3D y detecta estos cambios de gran escala con mayor precisión que las herramientas existentes, ofreciendo una vía distinta para estudiar genomas cancerosos y otras enfermedades.
Ver los cambios del genoma a través de mapas de contacto 3D
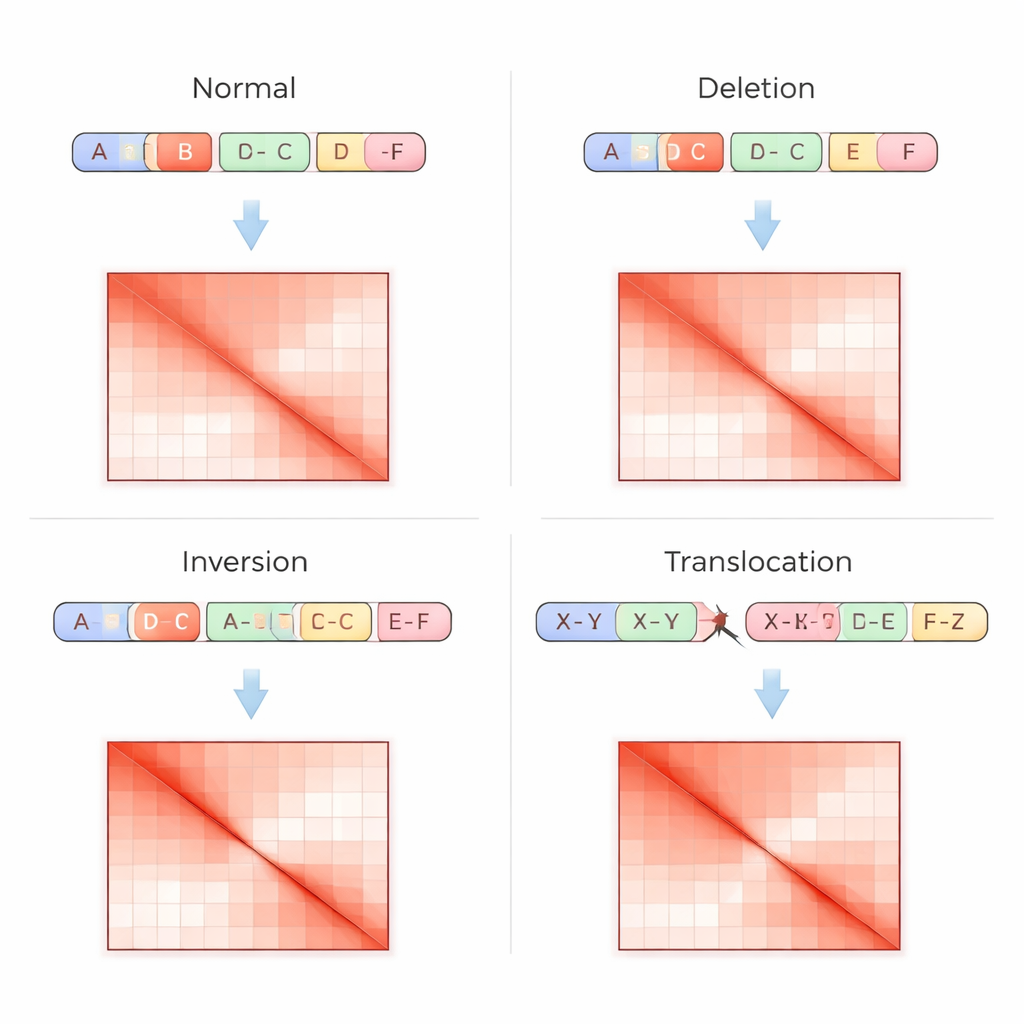
Las pruebas genómicas tradicionales leen el ADN como una secuencia lineal, lo que dificulta la detección de reordenamientos complejos, especialmente en regiones repetitivas o cuando los fragmentos se mueven sin cambiar su número de copia. La técnica Hi-C aborda el problema de forma diferente: mide con qué frecuencia partes distantes del ADN se tocan físicamente dentro del núcleo y registra estos contactos en una cuadrícula, o matriz de contactos, donde los puntos más brillantes indican interacciones más fuertes. Las variantes estructurales dejan huellas distintivas en estas matrices —como franjas ausentes cuando una región se ha eliminado, patrones especulares cuando un segmento se ha invertido, o puntos calientes fuera de la diagonal cuando dos cromosomas se han fusionado. VarHiCNet está diseñado para reconocer automáticamente esos patrones visuales.
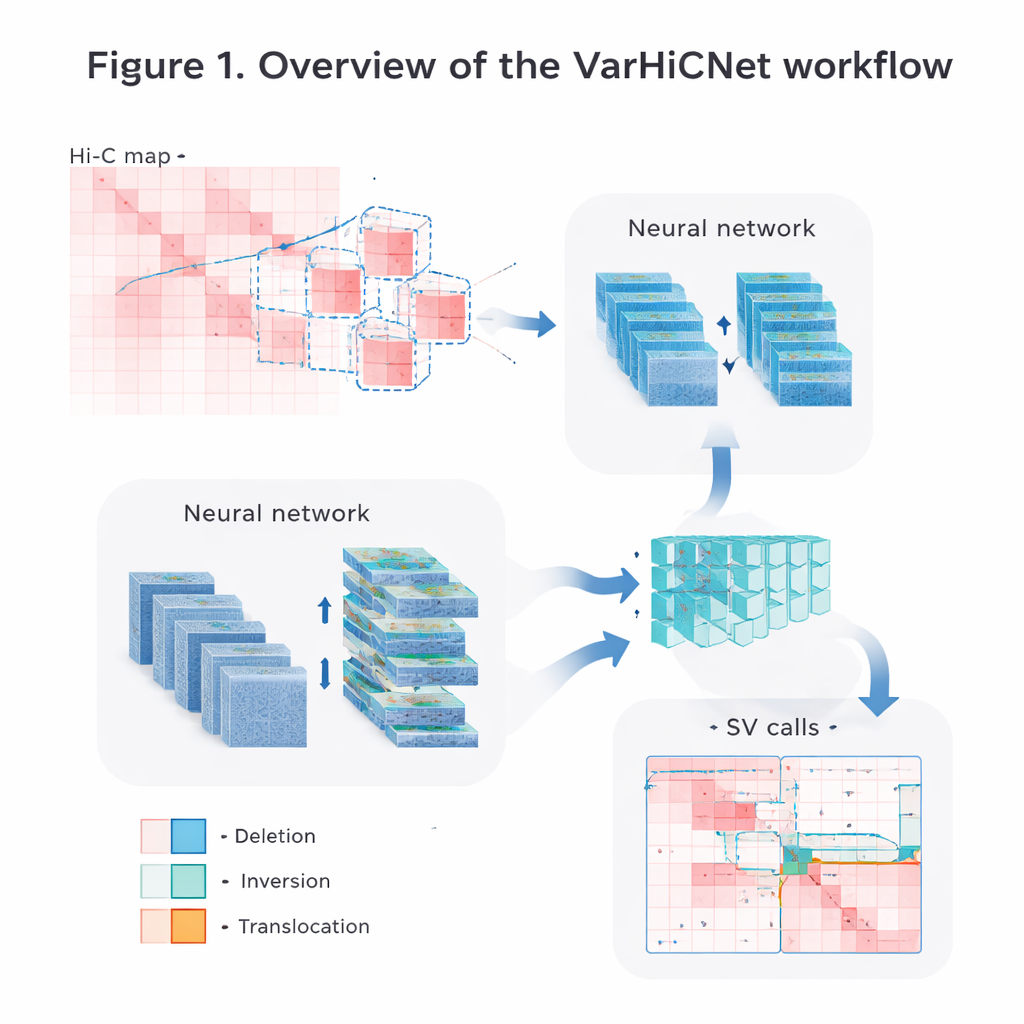
Convertir mapas genómicos en imágenes para IA
Los autores convierten los datos crudos de contactos Hi-C en imágenes que los sistemas de visión por computador pueden procesar con facilidad. Primero normalizan con cuidado las matrices para corregir la caída natural en la frecuencia de contactos a medida que los segmentos de ADN se separan, preservando a la vez las señales de interacción tanto cercanas como de largo alcance. Luego exploran cada cromosoma con ventanas cuadradas solapadas y recortan muchas submatrices más pequeñas. Cada submatriz se redimensiona a una imagen en color estandarizada de 800 por 800 píxeles, donde diferentes intensidades de contacto se traducen en tonos rojizos distribuidos en tres canales de color. Esta representación tipo imagen permite al modelo reutilizar técnicas potentes desarrolladas originalmente para reconocer objetos en fotografías.
Tomando prestadas estrategias de la detección de objetos
VarHiCNet trata cada variante estructural potencial como si fuera un “objeto” en una imagen. Se apoya en un marco moderno de detección de objetos llamado RT-DETR, que emplea una combinación de redes neuronales convolucionales y Transformers para resaltar regiones importantes. Un backbone ResNet extrae primero características multiescala: las capas superficiales mantienen el detalle fino necesario para localizar con precisión los puntos de ruptura, mientras que las capas más profundas capturan patrones más amplios que señalan eventos de gran tamaño. Un módulo de fusión de características mezcla después la información de varias capas para conservar tanto las pistas locales como las globales. Otro bloque personalizado, inspirado en el agrupamiento piramidal espacial (spatial pyramid pooling), ajusta cuánto del entorno ve el modelo a la vez, haciéndolo sensible a variantes que abarcan desde tramos relativamente pequeños hasta regiones muy extensas del ADN.
De regiones candidatas a tipos de variante precisos
Una vez que VarHiCNet ha propuesto regiones candidatas en la imagen Hi-C, debe refinarlas hasta obtener puntos de ruptura exactos y tipos específicos de variante, como deleciones, inversiones, duplicaciones o translocaciones. Para ello, el sistema hace un zoom en el vecindario alrededor de cada punto de ruptura predicho y reduce su complejidad usando una técnica matemática llamada análisis de componentes principales, que destaca dónde el patrón de contactos cambia con mayor brusquedad. Estas representaciones compactas se alimentan después a un clasificador basado en Transformers que aprende las diferencias sutiles en los patrones locales para cada categoría de variante. El resultado es una llamada detallada por cada evento: dónde ocurre en el genoma y qué tipo de cambio estructural representa.
Rendimiento en diversas líneas celulares tumorales
Los investigadores evaluaron VarHiCNet con datos Hi-C de seis líneas celulares humanas cancerosas diferentes, que cubren tumores de sangre, mama, cerebro, riñón, pulmón y próstata. Utilizando un catálogo de alta confianza de variantes estructurales conocidas como estándar de oro, compararon su método con varias herramientas líderes que también analizan datos Hi-C. Tanto para eventos dentro de un mismo cromosoma como entre cromosomas, VarHiCNet alcanzó en general puntuaciones F1 más altas o comparables, lo que indica un mejor equilibrio entre sensibilidad y precisión frente a otros enfoques. Fue especialmente eficaz en la detección de translocaciones balanceadas e inversiones —reordenamientos que a menudo dejan poca huella en la secuenciación estándar pero que muestran firmas claras en el plegamiento 3D. Los autores también demostraron que sus decisiones de diseño, como la resolución de la imagen y los módulos de fusión de características, mejoraron consistentemente el rendimiento en pruebas controladas.
Qué implica esto para entender las enfermedades
En términos cotidianos, VarHiCNet ofrece a los científicos una forma más inteligente de “ver” cómo se pliega el genoma en 3D y de detectar reordenamientos grandes relacionados con la enfermedad que podrían pasarse por alto con la secuenciación convencional. Al convertir mapas de contacto complejos en imágenes y aplicar redes neuronales modernas de estilo visión, el método puede detectar y clasificar con alta fiabilidad muchos tipos de variaciones estructurales en distintos tipos de cáncer. Aunque todavía presenta dificultades con cambios muy pequeños o altamente enmarañados y depende de datos de entrenamiento ricos, VarHiCNet apunta hacia un futuro en el que la arquitectura del genoma en 3D sea una parte habitual de cómo leemos, interpretamos y, eventualmente, dirigimos los cambios genéticos que subyacen al cáncer y a otras enfermedades.
Cita: Shen, J., Wang, H., Zhai, H. et al. A method for structural variant detection using Hi-C contact matrix and neural networks. Sci Rep 16, 7324 (2026). https://doi.org/10.1038/s41598-026-37678-6
Palabras clave: variación estructural, Hi-C, aprendizaje profundo, genómica del cáncer, genoma 3D