Clear Sky Science · es
Los pools de APOL1 en la membrana plasmática resisten la degradación proteica rápida
Por qué importa el “truco de desaparición” de una proteína renal
Una gran proporción de las formas graves de enfermedad renal en personas de ascendencia africana reciente se ha vinculado a dos variantes de un único gen, APOL1. Aun así, los científicos siguen sin explicar con precisión cómo este gen daña las células renales sin perjudicar a la mayoría de los portadores. Este estudio plantea una pregunta aparentemente simple con grandes implicaciones: una vez que la proteína APOL1 se sintetiza dentro de las células, ¿cuánto tiempo permanece y dónde es más estable? Las respuestas revelan una personalidad sorprendentemente dividida: APOL1 se destruye rápidamente dentro de las células, pero permanece obstinadamente estable cuando está incrustada en la superficie exterior de la célula, una pista que podría orientar futuras terapias.
Gen de riesgo con doble filo
El gen APOL1 ayuda a proteger a los humanos frente a ciertos parásitos, una ventaja evolutiva que probablemente explica por qué sus variantes de riesgo, denominadas G1 y G2, son comunes en poblaciones africanas. Desafortunadamente, las personas que heredan dos copias de estas variantes tienen un riesgo marcadamente mayor de sufrir trastornos renales agrupados como enfermedades renales mediadas por APOL1. Trabajos previos mostraron que cuando aumentan los niveles de APOL1—con frecuencia en respuesta a la inflamación—la proteína puede volverse tóxica, especialmente en las delicadas células filtradoras renales conocidas como podocitos. Pero la mayoría de los estudios se centraron en qué activa APOL1. Se sabía mucho menos sobre cómo las células la desactivan, por ejemplo mediante su degradación.
Rastreando una proteína frágil dentro de las células
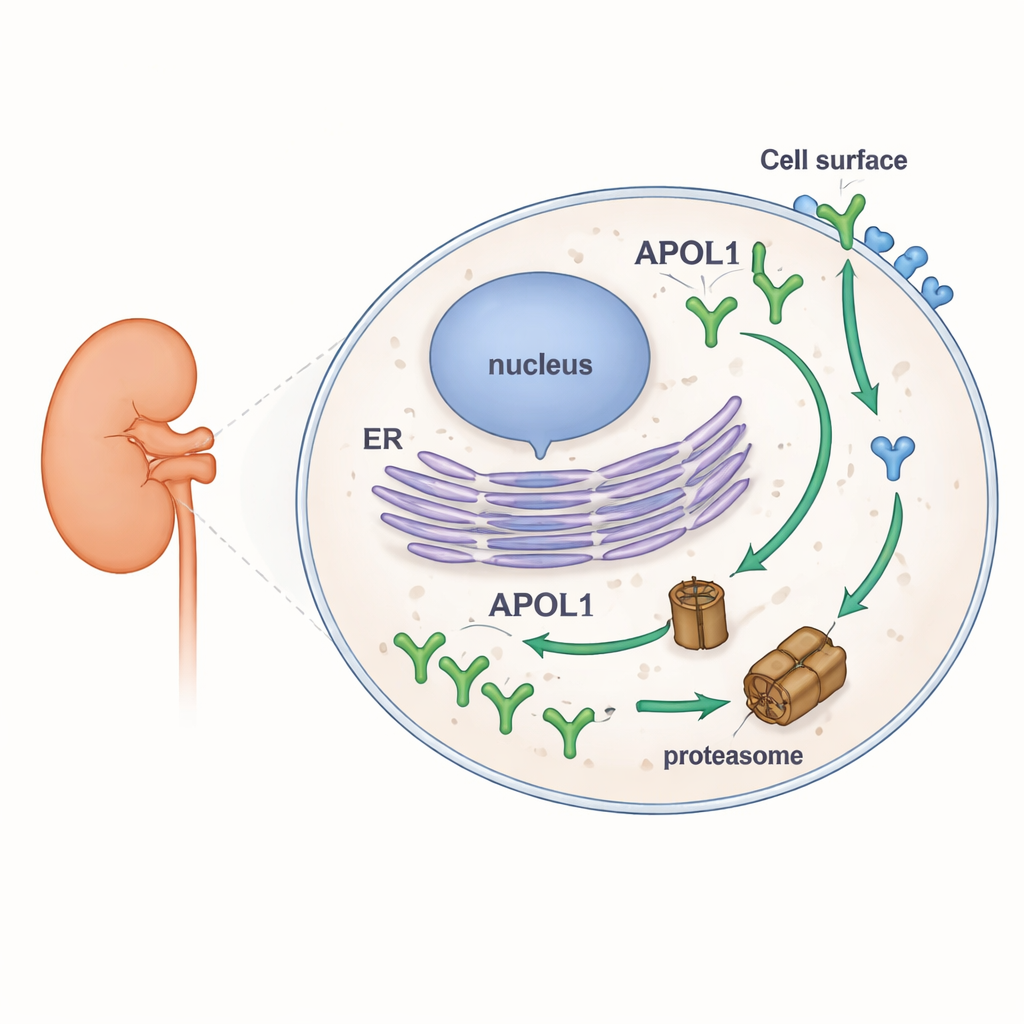
Para explorar la estabilidad de APOL1, los investigadores diseñaron líneas celulares humanas que producían versiones marcadas con fluorescencia de APOL1 y de su pariente más cercano, APOL2. Esto les permitió observar cuánto de cada proteína se acumulaba o desaparecía bajo distintas condiciones mediante Western blot, microscopía y citometría de flujo. Bloquearon la principal maquinaria degradadora de proteínas de la célula, el proteasoma, y por separado detuvieron la síntesis de nuevas proteínas. Cuando se inhibieron los proteasomas, los niveles de APOL1 aumentaron rápidamente, lo que muestra que normalmente se descompone a gran velocidad. Cuando se detuvo la síntesis de proteínas nuevas, los niveles de APOL1 cayeron con rapidez. En marcado contraste, APOL2 apenas cambió bajo ninguno de los dos tratamientos, revelándola como una proteína mucho más estable. Es importante destacar que la alta rotación de APOL1 fue la misma para la versión normal (G0) y para las variantes de riesgo renal (G1 y G2), y se mantuvo en varias formas naturales de APOL1 que difieren en su disposición en las membranas.
Pistas en la secuencia y la historia de dos vecindarios
Al indagar en la estructura de la proteína, el equipo utilizó herramientas informáticas para escanear APOL1 y APOL2 en busca de segmentos flojos y no estructurados conocidos como regiones intrínsecamente desordenadas. Tales regiones a menudo actúan como señales de “cómeme” para el proteasoma. Identificaron dos regiones candidatas fuertes en APOL1 que estaban en gran medida ausentes en APOL2. Para probar si el extremo anterior único de APOL1 contribuye a su fragilidad, crearon híbridos: una APOL1 acortada que carecía de sus primeros 59 aminoácidos, y una quimera de APOL2 que portaba ese segmento de APOL1. Añadir el fragmento N-terminal de APOL1 a APOL2 hizo que APOL2 se degradara más rápido, mientras que la APOL1 truncada siguió siendo inestable, lo que sugiere que más de una parte de APOL1 favorece su descomposición rápida. En conjunto, estos resultados vinculan los inusuales segmentos flexibles de APOL1 con su alta rotación, sin ligar este comportamiento específicamente a las variantes causantes de la enfermedad.
Proteína obstinada en la superficie celular
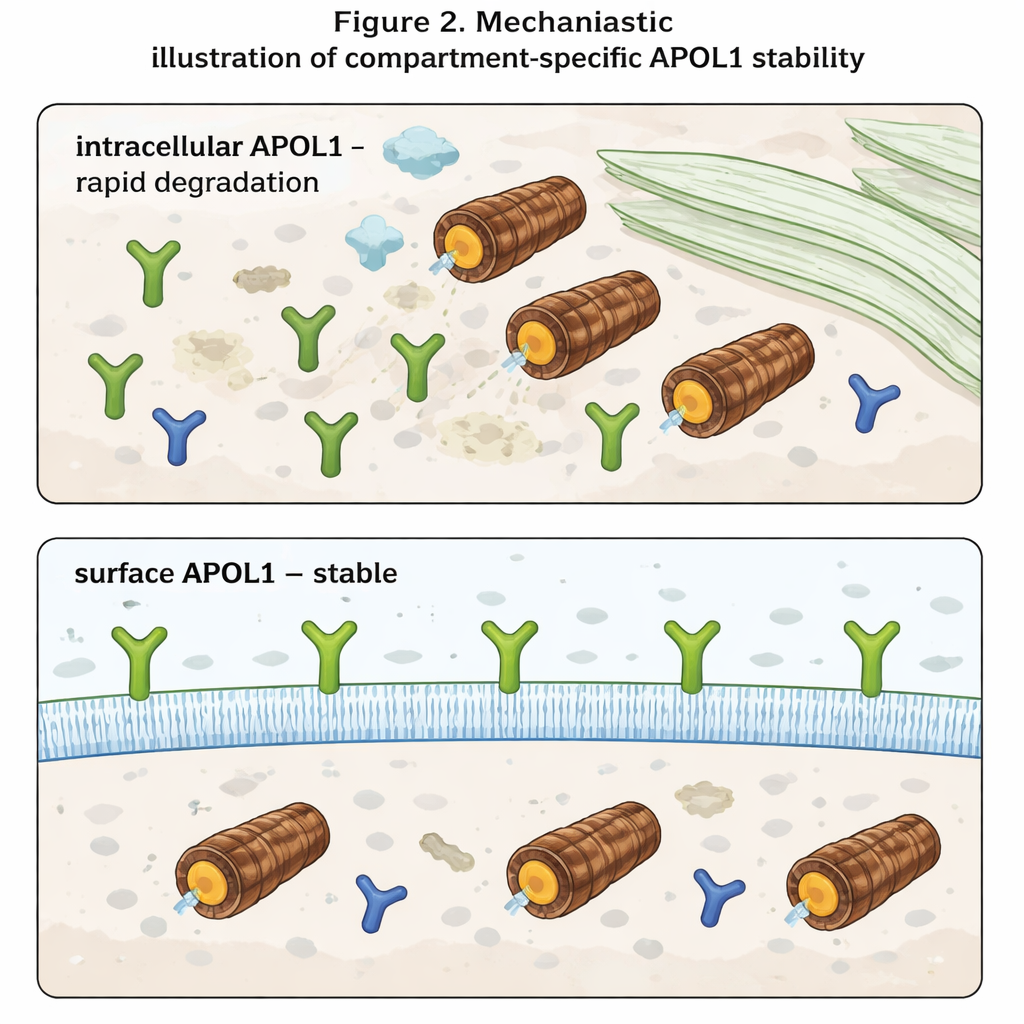
El hallazgo más llamativo surgió cuando los autores distinguieron entre APOL1 dentro de la célula y APOL1 en la superficie celular. Utilizando anticuerpos que reconocen solo la porción de APOL1 expuesta al exterior, midieron los niveles superficiales por separado de los niveles totales. Dentro de la célula, APOL1 se comportó como cabía esperar: se acumuló cuando se bloquearon los proteasomas y desapareció rápidamente cuando se detuvo la nueva síntesis. La APOL1 superficial, sin embargo, apenas se movió bajo cualquiera de las condiciones. Una vez que las moléculas de APOL1 llegaron a la membrana plasmática, demostraron ser altamente resistentes a la degradación rápida. Además, aunque las variantes de riesgo producían menos APOL1 total que la versión normal, sus niveles en la superficie eran similares. Esto sugiere que las APOL1 de riesgo y las normales se eliminan a ritmos comparables dentro de la célula, pero que los pools embebidos en la membrana—que se piensa que forman canales iónicos y que impulsan la toxicidad—se preservan en todas las variantes.
Qué significa esto para tratamientos futuros
Para el público no especializado, la conclusión es que APOL1 se comporta de forma muy diferente según dónde se encuentre. Dentro de la célula es una proteína de corta vida, rápidamente reconocida y degradada. En la superficie celular se vuelve duradera y relativamente protegida, incluso cuando se altera la maquinaria degradadora de la célula. Dado que la enfermedad parece surgir cuando los canales de APOL1 en la superficie alteran el equilibrio de iones como sodio y potasio, las terapias podrían necesitar centrarse menos en los niveles totales de APOL1 y más en cuánto llega y persiste en la membrana plasmática. Estrategias que reduzcan el tráfico de APOL1 hacia la superficie o que desestabilicen selectivamente la fracción superficial podrían, en principio, mitigar el daño renal sin bloquear por completo las funciones inmunitarias beneficiosas del gen.
Cita: Höffken, V., Alvermann, L., Niggemeier, D. et al. APOL1 plasma membrane pools resist rapid protein degradation. Sci Rep 16, 6718 (2026). https://doi.org/10.1038/s41598-026-37647-z
Palabras clave: APOL1, enfermedad renal, degradación de proteínas, membrana plasmática, proteasoma