Clear Sky Science · es
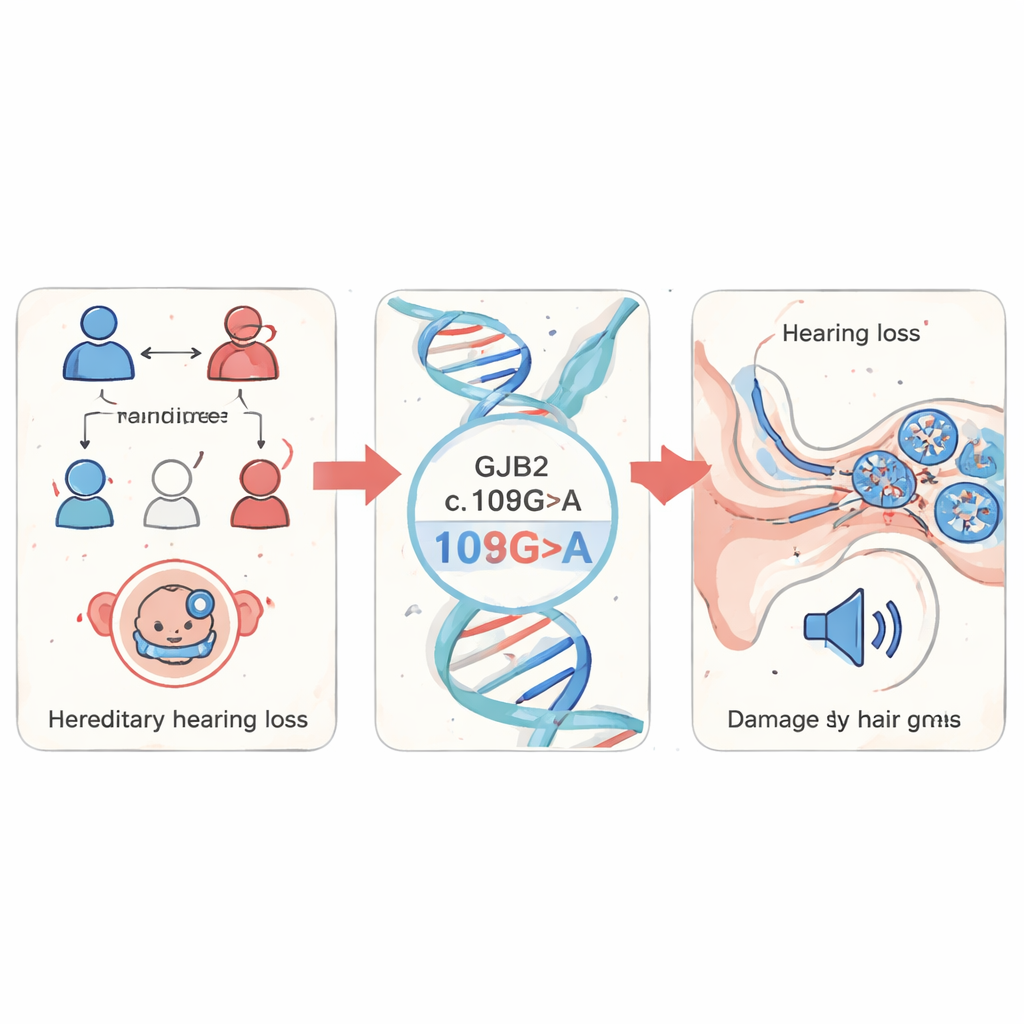
Mutación GJB2 c.109G>A que activa la vía de apoptosis mitocondrial mediada por IFI27 y conduce a hipoacusia hereditaria no sindrómica
Por qué las diminutas células del oído importan para el futuro de los niños
La hipoacusia presente al nacer afecta a millones de niños en todo el mundo y con frecuencia determina cómo aprenden a hablar, rinden en la escuela y se relacionan con los demás. Uno de los culpables genéticos más comunes es el gen GJB2, pero los médicos no comprendían por completo cómo las alteraciones en este gen dañan el oído interno. Este estudio usa pez cebra y células humanas para trazar la cadena de eventos desde un único cambio de ADN en GJB2 hasta la muerte de las delicadas células sensoriales del sonido, y señala a una molécula nueva, IFI27, como posible diana para futuros tratamientos.
Un cambio genético común detrás del silencio infantil
Los investigadores comenzaron cribando muestras de sangre de 1.199 niños con sospecha de hipoacusia hereditaria en la provincia de Fujian, China. Se centraron en varios genes de sordera bien conocidos y hallaron que las alteraciones en GJB2 dominaban el panorama, representando el 85% de todas las mutaciones detectadas. Entre ellas, un cambio específico denominado c.109G>A (también conocido como p.Val37Ile) fue el más frecuente. Esta variante es relativamente común en la población general pero está fuertemente enriquecida en personas con hipoacusia, lo que sugiere que desempeña un papel importante en la hipoacusia no sindrómica —problemas auditivos que ocurren sin otras afecciones médicas.
Siguiendo el daño en un pez transparente
Para ver qué hace esta mutación en un organismo vivo, el equipo recurrió al pez cebra, un pequeño pez de agua dulce cuyos embriones son transparentes y comparten muchos genes y estructuras del oído con los humanos. Modificaron embriones de pez cebra para que produjeran ya fuera GJB2 humano normal o la versión mutante c.109G>A, y también usaron un enfoque de “knockdown” para reducir el gen gjb2 propio del pez. Los embriones con el gen mutante o reducido mostraron crecimiento retrasado, colas curvadas e hinchazón alrededor del corazón, signos de un desarrollo alterado. Lo más importante, sus oídos internos eran claramente anormales: estructuras clave llamadas otolitos eran más pequeñas y estaban más separadas, y la región coclear llena de líquido estaba reducida. Cuando los científicos reintrodujeron GJB2 normal junto con el mutante, muchos de estos problemas estructurales mejoraron, lo que demuestra que la mutación por sí misma causaba los defectos.
De oídos defectuosos a un comportamiento auditivo peor
Dado que la audición depende de diminutas “células ciliadas” que convierten las vibraciones sonoras en señales nerviosas, el equipo tiñó estas células en el pez cebra. Los peces con la mutación GJB2 o con el knockdown tenían muchas menos células ciliadas tanto en el oído interno como a lo largo de la superficie corporal, donde el pez cebra también percibe el movimiento del agua. Los investigadores midieron luego la respuesta de los peces al sonido. Usando un sistema automatizado de seguimiento, registraron cuánto y qué tan rápido nadaban larvas de 5 días al exponerse a breves impulsos sonoros. Los peces normales y los que expresaban GJB2 salvaje reaccionaron nadando más y más rápido, mientras que los peces mutantes y con knockdown apenas cambiaron su comportamiento, indicando una audición deteriorada. De nuevo, añadir GJB2 normal restauró en parte tanto el número de células ciliadas como el movimiento inducido por el sonido.
Una vía de muerte dentro de las centrales energéticas de la célula
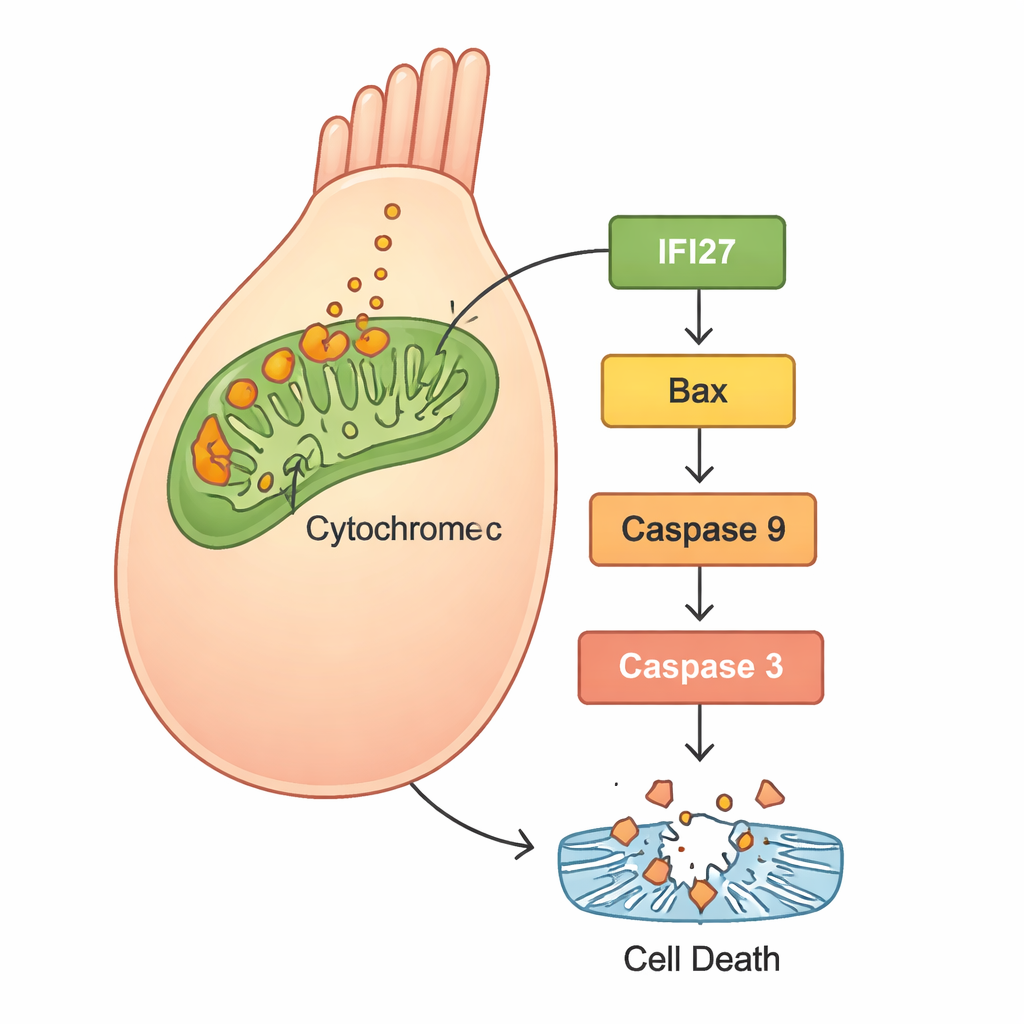
Para entender qué ocurría dentro de las células, los científicos utilizaron secuenciación de ARN para comparar la actividad génica entre pez cebra normal y aquellos con gjb2 reducido. Se activó con fuerza un conjunto de genes ligado a la “vía de apoptosis mitocondrial”, una ruta de autodestrucción centrada en las fábricas de energía de la célula. En particular, destacaron varios miembros de la familia IFI27, junto con actores bien conocidos de la muerte celular como Bax, citocromo c, Apaf1 y caspasas. Experimentos de seguimiento en células humanas HEK293 confirmaron el patrón: las células con GJB2 mutante produjeron más especies reactivas de oxígeno (ROS, una forma de estrés oxidativo), liberaron más citocromo c desde las mitocondrias y activaron proteínas de apoptosis, lo que condujo a un aumento de la muerte celular. Cuando los investigadores silenciaron IFI27 en células portadoras del gen mutante, los niveles de ROS descendieron, las señales de muerte se atenuaron y menos células experimentaron apoptosis.
Qué significa esto para futuros tratamientos
En conjunto, los hallazgos sugieren una historia clara: la mutación GJB2 c.109G>A altera el desarrollo y la función del oído interno, no solo al modificar la comunicación celular, sino también al desencadenar estrés mitocondrial. Este estrés aumenta IFI27 y genes relacionados, libera citocromo c y activa una cascada de proteínas que empujan a las células ciliadas hacia la muerte programada. Debido a que las células ciliadas no se regeneran fácilmente en humanos, su pérdida conduce a déficits auditivos permanentes. Al demostrar que reducir IFI27 puede mitigar esta cascada destructiva en células humanas, el estudio destaca a IFI27 como una diana prometedora para fármacos o terapias génicas. Aunque tales tratamientos están aún lejos en el futuro —y probablemente deberán administrarse muy temprano en la vida—, este trabajo ofrece una hoja de ruta molecular concreta para convertir una mutación génica antes misteriosa en una causa potencialmente prevenible de sordera infantil.
Cita: Chen, Y., Zhao, P., Lin, Q. et al. GJB2 c.109G > A mutation activating IFI27-mediated mitochondrial apoptosis pathway leading to hereditary non-syndromic hearing loss. Sci Rep 16, 6240 (2026). https://doi.org/10.1038/s41598-026-37393-2
Palabras clave: pérdida auditiva genética, mutación GJB2, modelo en pez cebra, apoptosis mitocondrial, IFI27