Clear Sky Science · es
Selección de características adaptativa guiada por clústeres difusos: simple, rápida y eficiente para datos microarray bioinformáticos binarios de alta dimensión y fuertemente desequilibrados
Por qué esto importa en la investigación génica
Las pruebas modernas de expresión génica pueden medir decenas de miles de genes en una sola muestra de paciente. Ese aluvión de datos promete un diagnóstico de cáncer más temprano y mejores decisiones de tratamiento, pero también crea un problema: la mayoría de esos genes son ruidosos, redundantes o están asociados principalmente a los casos comunes, no a los raros y peligrosos. Este artículo presenta una nueva forma de cribar conjuntos masivos de datos de expresión génica para que los ordenadores puedan identificar de forma fiable a pacientes pertenecientes a una pequeña minoría difícil de detectar usando solo un conjunto reducido y cuidadosamente seleccionado de genes.
El desafío de demasiados genes muy parecidos
Los experimentos de microarrays suelen registrar miles de niveles de actividad génica para apenas unos cientos de pacientes. Normalmente, una clase (por ejemplo, un subtipo de cáncer común) supera con creces a la otra, generando datos fuertemente desequilibrados. En este escenario, muchos genes se comportan de maneras muy similares y los patrones de pacientes de la mayoría y de la minoría pueden solaparse. Los métodos de aprendizaje estándar tienden a centrarse en la clase mayoritaria y se confunden con genes redundantes, lo que conduce a sobreajuste y a una detección deficiente de subtipos raros. Los métodos tradicionales de reducción de dimensionalidad o bien sacrifican la interpretabilidad al construir nuevas características mixtas, o seleccionan genes sin evaluar detenidamente cuánto ayudan a un clasificador a reconocer los casos minoritarios.
Una nueva hoja de ruta para una selección génica más inteligente
Los autores introducen AFCG‑SFE, un modelo adaptativo de selección de características diseñado específicamente para datos de expresión génica de alta dimensión y desequilibrados. El método parte de una búsqueda simple de “agente único” que activa o desactiva genes y prueba cuánto respaldan la clasificación, pero la enriquece con varios pasos impulsados por los datos. Primero, agrupa los genes según cómo se comportan de forma similar y permite que los genes pertenezcan a más de un grupo para reflejar la realidad biológica de que un gen puede intervenir en múltiples vías. Dentro de cada grupo, ordena los genes según cuánta información aportan sobre la etiqueta de la enfermedad y conserva solo unos pocos representantes clave, reduciendo drásticamente la redundancia antes de que comience la búsqueda principal. 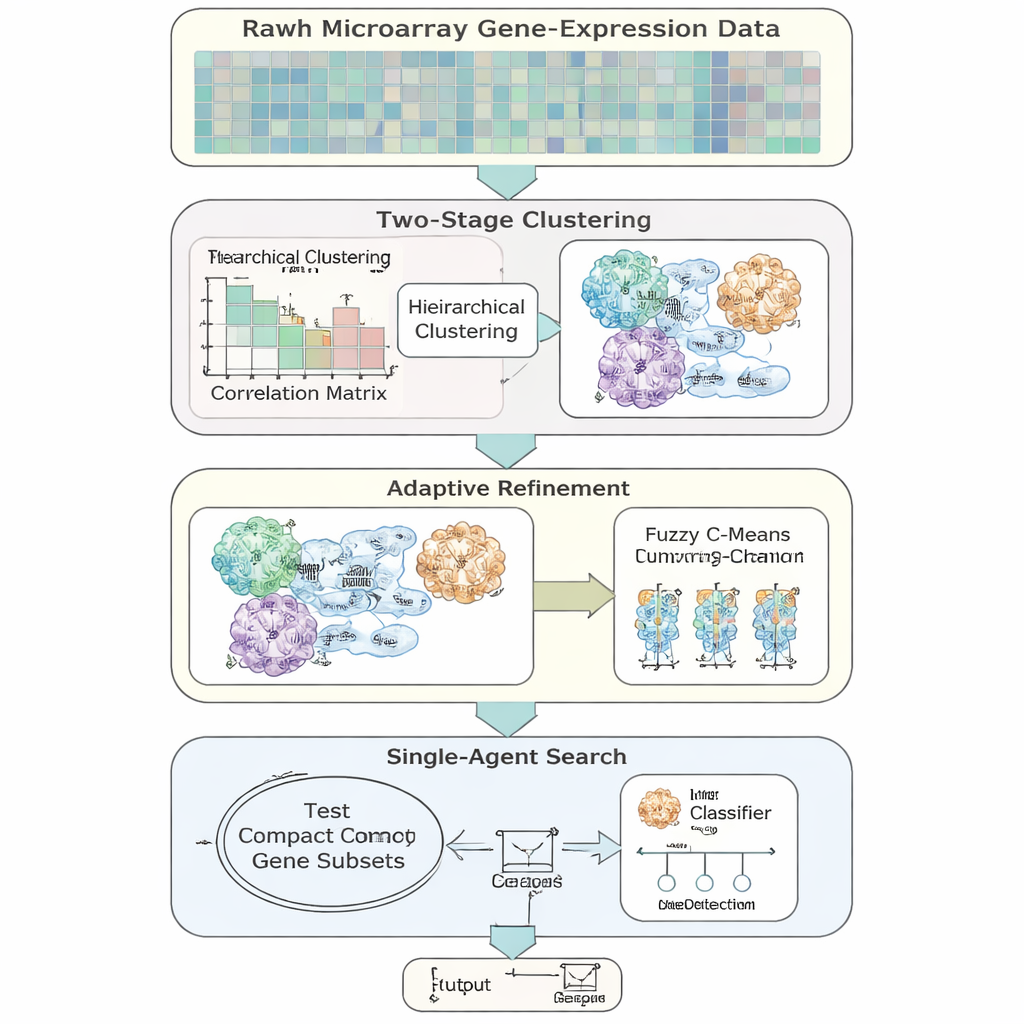
Hacer que el ordenador valore a los pacientes raros
En lugar de centrarse en la precisión simple, AFCG‑SFE usa una puntuación de aptitud que enfatiza métricas adecuadas para datos sesgados, incluyendo el equilibrio entre identificar correctamente casos minoritarios y mayoritarios y el rendimiento a lo largo de todos los umbrales de decisión. La función de aptitud también incluye penalizaciones por seleccionar demasiados genes o muchos genes del mismo clúster, y una recompensa para los genes que muestran fuerte dependencia con la etiqueta de la enfermedad. Es importante que la intensidad de estas penalizaciones y recompensas se ajuste automáticamente a partir de propiedades del conjunto de datos, como cuántos genes hay por paciente y cuánto se solapan las clases, en vez de requerir ajuste manual. Esto hace que el método sea más robusto y más fácil de transferir entre estudios.
Adaptarse a la dificultad del problema
Una idea clave es que el algoritmo no debe buscar siempre el conjunto de genes más pequeño posible. Cuando las dos clases son muy difíciles de separar o están fuertemente solapadas, el método eleva automáticamente un umbral inferior sobre cuántos genes deben conservarse, asegurando que señales raras pero importantes no se descarten. A medida que avanza la búsqueda, AFCG‑SFE ajusta gradualmente un límite por clúster sobre cuántos genes pueden sobrevivir en cada grupo, respetando a la vez ese mínimo. El resultado es un panel compacto y diverso de genes que captura la estructura de los datos sin estar dominado por un único patrón redundante. 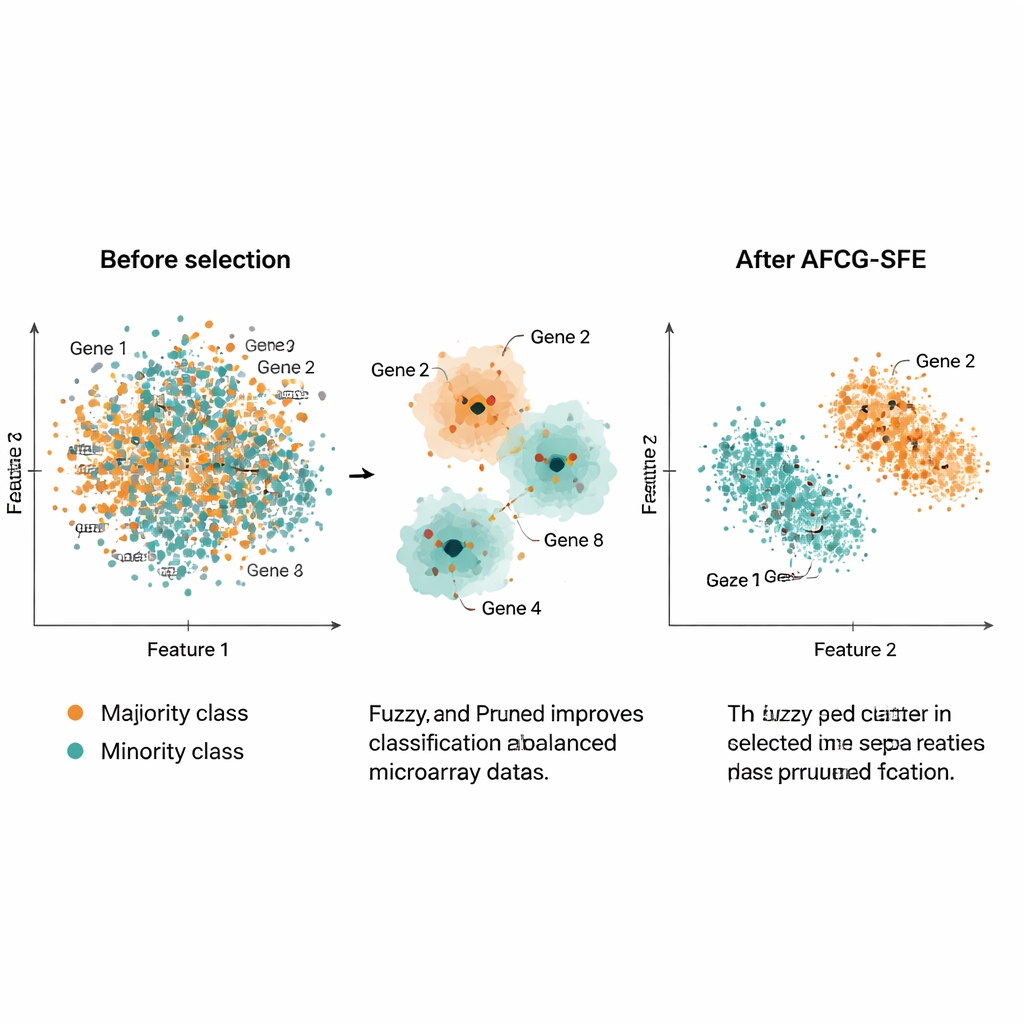
Qué muestran los experimentos
Los autores evaluaron AFCG‑SFE en 20 conjuntos públicos de microarrays de cáncer, cada uno con miles de genes pero solo alrededor de 100–200 muestras y un fuerte desequilibrio de clases. Compararon su método con varias búsquedas evolutivas de referencia, filtros simples y enfoques integrados que incorporan la selección de características en el clasificador. En una batería de medidas —incluyendo F‑measure, precisión balanceada, área bajo la curva ROC y una métrica de sobreajuste— AFCG‑SFE fue el mejor o empató como el mejor en todos los conjuntos. Típicamente seleccionó menos de 25 genes (a menudo tan pocos como 6–8), eliminando más del 99% de las características originales y, aun así, mejorando o manteniendo el rendimiento de la clasificación. También redujo un índice de complejidad que captura cuánto se solapan las clases en el espacio de características, indicando una separación más clara tras la selección.
Conclusión para no especialistas
En términos prácticos, este trabajo ofrece una forma de reducir perfiles enormes y ruidosos de expresión génica a conjuntos muy pequeños de genes informativos que aún permiten a los ordenadores reconocer con precisión subgrupos de pacientes raros. Al agrupar inteligentemente genes similares, premiar a aquellos que realmente siguen la enfermedad y proteger explícitamente contra el sesgo hacia la clase mayoritaria, AFCG‑SFE aporta tanto mejores predicciones como paneles génicos mucho más simples. Esa combinación puede ayudar a los investigadores a identificar biomarcadores potenciales, diseñar pruebas diagnósticas más interpretables y, en última instancia, mejorar cómo las herramientas de medicina de precisión funcionan con datos biológicos reales e imperfectos.
Cita: Tye, Y.W., Chew, X., Yusof, U.K. et al. Adaptive fuzzy cluster-guided simple, fast, and efficient feature selection for high-dimensional and highly imbalanced binary-class bioinformatics microarray data. Sci Rep 16, 6650 (2026). https://doi.org/10.1038/s41598-026-37086-w
Palabras clave: expresión génica, selección de características, datos desequilibrados, microarray, subtipos de cáncer