Clear Sky Science · es
rhinotypeR permite la asignación reproducible de genotipos de rinovirus a partir de secuencias VP4/2
Por qué siguen importando esos pequeños virus del resfriado
La mayoría de nosotros consideramos el resfriado común más una molestia que una amenaza grave. Sin embargo, los virus que causan muchos resfriados —los rinovirus humanos— también se asocian con infecciones pulmonares graves, crisis de asma y exacerbaciones de enfermedades pulmonares crónicas. Para seguir cómo evolucionan y se propagan estos virus, los científicos necesitan clasificarlos en “tipos” genéticos precisos, como si pusieran códigos de barras a los productos. Este artículo presenta rhinotypeR, un paquete de software gratuito y de código abierto que hace que este etiquetado genético sea más exacto, coherente y fácil de repetir, ayudando a los equipos de salud pública a vigilar con más claridad una familia de virus respiratorios a menudo subestimada.
La variedad oculta en los resfriados comunes
Los rinovirus humanos son extraordinariamente frecuentes, apareciendo en hasta el 60 % de las muestras de personas con enfermedad respiratoria aguda. Lejos de ser un único virus, se dividen en tres grupos principales, denominados A, B y C, y al menos 169 tipos genéticos reconocidos. Los distintos tipos se comportan de manera distinta: algunos se vinculan con más frecuencia a infecciones graves en niños y a crisis de asma, mientras que otros se observan con menos frecuencia en enfermedad grave. Dado que estos tipos evolucionan de forma independiente y presentan características de superficie distintas, los científicos necesitan métodos fiables para diferenciarlos si quieren seguir cómo se desplazan los brotes por escuelas, hogares y comunidades.
De herramientas dispersas a un camino claro
Hasta ahora, asignar un tipo de rinovirus a partir de su código genético había sido un trabajo fragmentado. Los investigadores suelen centrarse en un tramo corto del genoma del virus llamado región VP4/2, alinearlo con cepas de referencia conocidas, medir cuánto difieren las secuencias y luego aplicar valores umbral para decidir a qué tipo pertenece cada muestra. Pero estos pasos se realizaban con una mezcla de programas, ediciones manuales y criterio personal. Eso dificultaba comparar o reproducir distintos estudios, incluso cuando usaban datos similares. rhinotypeR se creó específicamente para convertir este proceso multinivel y propenso a errores en un flujo de trabajo único y scriptado que cualquiera puede ejecutar y compartir.
Qué hace realmente el nuevo software
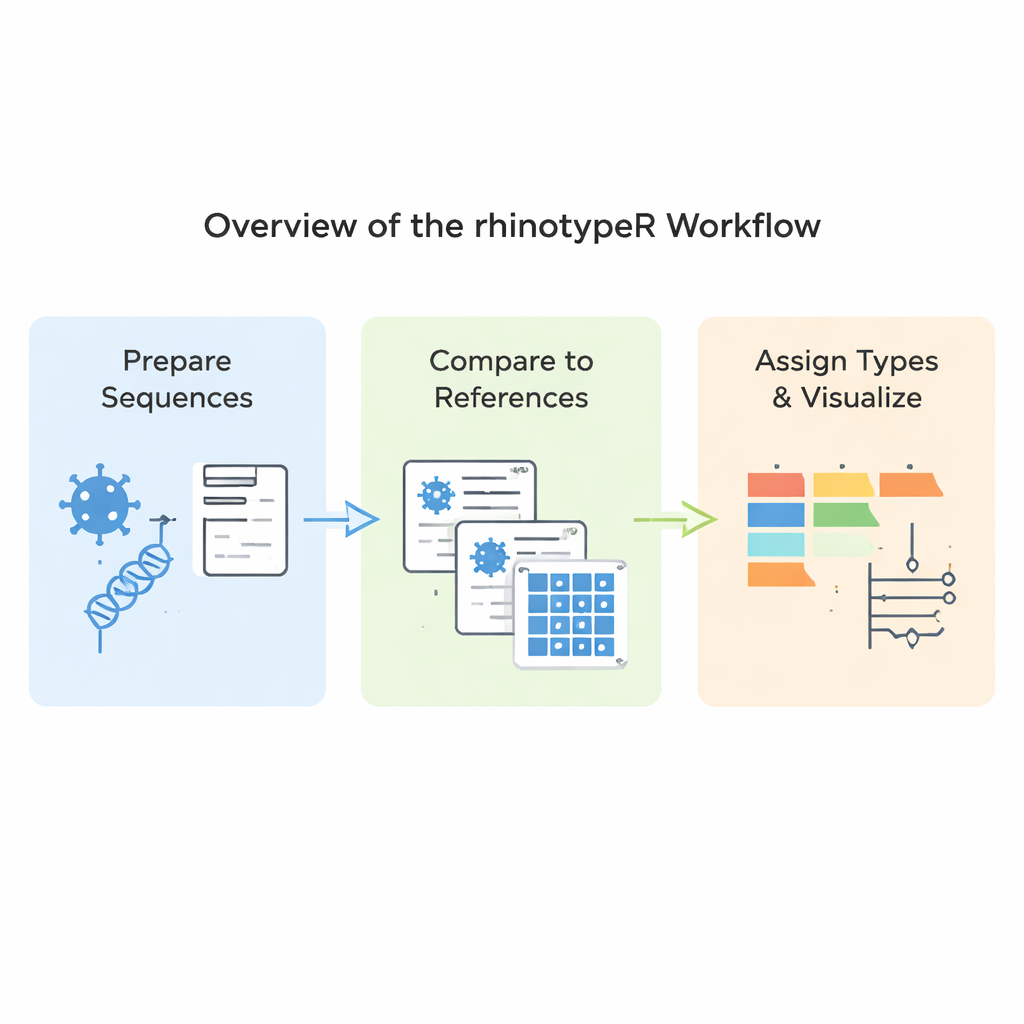
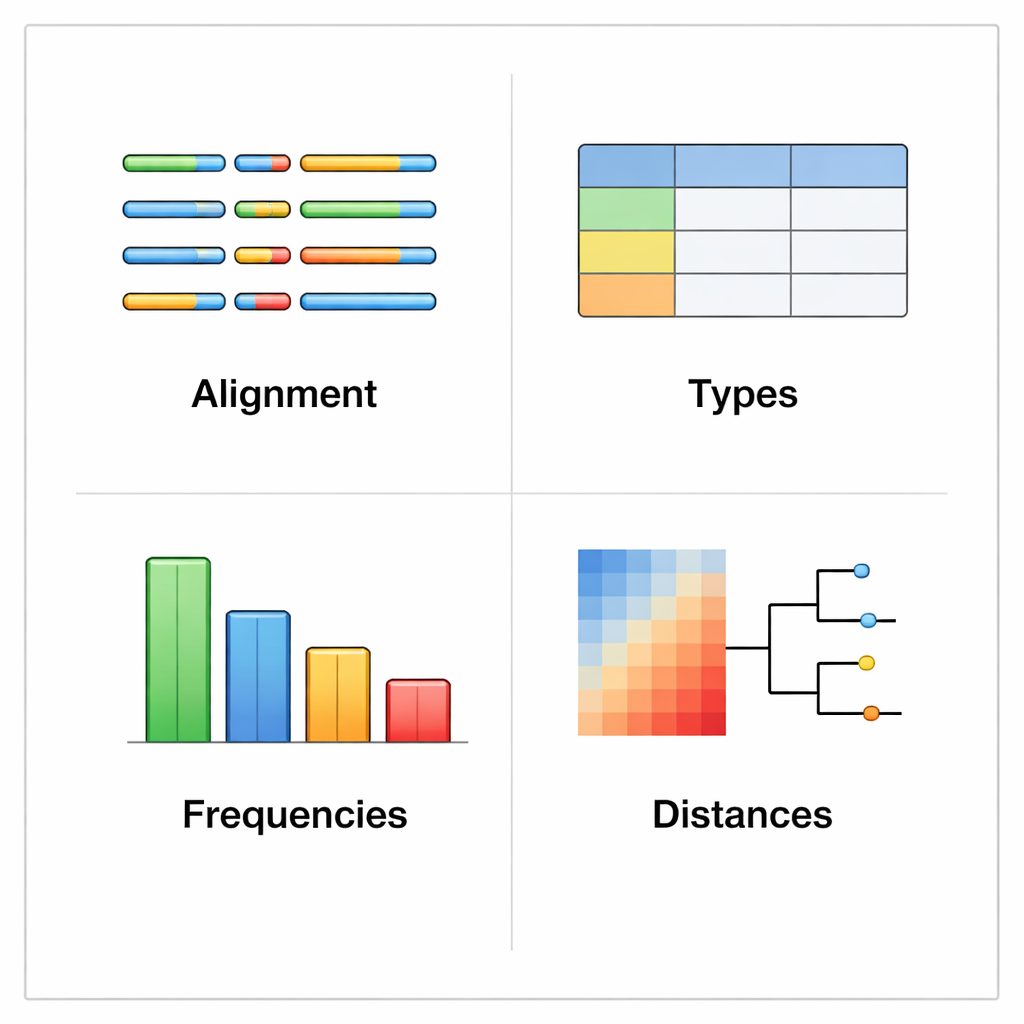
rhinotypeR se ejecuta dentro del entorno ampliamente usado R y Bioconductor para el análisis de datos. Toma un conjunto de secuencias VP4/2 de rinovirus y las procesa en tres etapas principales: preparar y alinear las secuencias, calcular la distancia de cada una respecto a un conjunto curado de tipos de referencia y, a continuación, asignar cada muestra al tipo conocido más cercano o marcarla como “no asignada” si es demasiado distinta. La misma herramienta puede producir salidas visuales, incluidos mapas en color de las diferencias genéticas, árboles filogenéticos sencillos y gráficos que muestran la prevalencia de cada tipo en un conjunto de datos. Los usuarios pueden alinear sus datos con programas externos si lo prefieren, o dejar que rhinotypeR gestione todo el proceso dentro de R para máxima reproducibilidad.
Poniendo la herramienta a prueba
Para comprobar que rhinotypeR ofrece resultados fiables, los autores compararon sus medidas de distancia con las de dos programas establecidos, ape y MEGA X, usando los mismos archivos de entrada y modelos. Los resultados coincidieron casi perfectamente; las pequeñas discrepancias se debieron al redondeo normal en la aritmética de ordenador, no a diferencias reales en el método. El equipo ejecutó luego rhinotypeR en una amplia colección de más de 2.300 secuencias de rinovirus de múltiples estudios previos, que abarcan más del 90 % de los tipos conocidos. En aproximadamente cuatro de cada cinco casos, la nueva herramienta coincidió exactamente con las etiquetas de tipo previas. La mayoría de los desacuerdos ocurrieron justo alrededor de los puntos de corte preestablecidos usados para separar un tipo de otro, que es precisamente donde se esperan las llamadas limítrofes. Importante, las muestras que no pudieron asignarse con confianza a un tipo conocido no mostraron signos de ser simplemente muestras de baja calidad o con poco virus, lo que sugiere que pueden reflejar una diversidad viral genuina.
Por qué esto importa para la salud pública
Para quienes no son especialistas, el mensaje clave es que rhinotypeR no reinventa la forma en que los científicos clasifican los virus del resfriado; en cambio, hace ese proceso más claro, transparente y fácil de reproducir. Al agrupar alineamiento, cálculos de distancia y asignación de tipos en un paquete de código abierto —junto con resúmenes visuales claros— ayuda a investigadores y programas de vigilancia a procesar miles de muestras de forma coherente. Esa coherencia mejora nuestra capacidad para comparar estudios de distintos lugares y tiempos, detectar de forma temprana linajes virales inusuales o emergentes y vincular patrones genéticos con tendencias de enfermedad en el mundo real. A largo plazo, herramientas como rhinotypeR fortalecen la monitorización rutinaria de resfriados aparentemente ordinarios que, en muchas personas, pueden desencadenar enfermedades graves.
Cita: Luka, M.M., Nanjala, R., Rashed, W.M. et al. rhinotypeR enables reproducible rhinovirus genotype assignment from VP4/2 sequences. Sci Rep 16, 6149 (2026). https://doi.org/10.1038/s41598-026-37050-8
Palabras clave: genotipado de rinovirus, vigilancia molecular, secuenciación VP4/2, herramientas bioinformáticas, virus respiratorios