Clear Sky Science · es
Evaluación comparativa de los métodos de perfilado transcriptómico dirigido HTG y TempO-Seq
Por qué esto importa para la atención del cáncer
Cuando médicos y científicos estudian el cáncer, a menudo recurren a las “moléculas mensajeras” de la célula —el ARN— para ver qué genes están activos o silenciados. Estos patrones pueden revelar cómo se comporta un tumor y qué tratamientos podrían ser más eficaces. Pero la mayoría de las muestras hospitalarias se almacenan en bloques de parafina tras el tratamiento con formalina, lo que daña el ARN, que es frágil. Este estudio plantea una pregunta práctica con grandes consecuencias para la investigación oncológica: ahora que una prueba de ARN muy utilizada ha desaparecido del mercado, ¿puede un método más reciente suplirla y ofrecer resultados igual de útiles a partir de estas muestras rutinarias preservadas?
Dos herramientas para leer la actividad génica
Durante años, muchos laboratorios confiaron en un método llamado HTG EdgeSeq Human Transcriptome Panel (HTP) para leer la actividad génica directamente a partir de pequeñas raspaduras de tejido fijado en formalina e incluido en parafina (FFPE). Este enfoque podía abarcar casi todos los genes humanos sin necesidad de extraer ARN previamente, ahorrando tiempo y preservando material valioso. Sin embargo, la empresa detrás de HTG EdgeSeq quebró, dejando a los investigadores buscando una alternativa. Una tecnología más reciente, TempO-Seq (TOS), de otro fabricante, promete capacidades similares: también dirige la detección a muchos genes a la vez, funciona con ARN dañado de muestras FFPE y está diseñada para ser sensible, reproducible y relativamente asequible.
Poniendo a prueba los métodos
El equipo de investigación comparó estas dos tecnologías cara a cara en un entorno muy práctico. Analizaron 21 muestras almacenadas de cáncer de endometrio, junto con tres materiales de referencia estándar de ARN, primero con HTG HTP y luego con TempO-Seq. Ambos métodos utilizaron paneles dirigidos que en conjunto cubrían más de 18.000 genes coincidentes. Los científicos aplicaron controles de calidad estrictos, asegurando que cada muestra produjera suficientes lecturas de secuenciación y que las mediciones fueran estables. También emplearon herramientas estadísticas para eliminar los "efectos de lote" —diferencias artificiales que pueden surgir simplemente porque las pruebas se realizaron en días, máquinas o plataformas distintos.
Qué coincide y qué no
Cuando el equipo examinó la expresión de genes individuales uno por uno, los dos métodos no siempre coincidieron. Las diferencias en cómo cada tecnología diseña sus sondas, prepara las muestras y cuenta las lecturas pueden hacer que las comparaciones de un solo gen sean ruidosas. Sin embargo, este panorama cambió al analizar patrones más amplios que combinan información de muchos genes a la vez. Las firmas multigénicas —como las usadas para agrupar tumores en subtipos moleculares, estimar cuántas células inmunitarias hay en una muestra o evaluar la pureza del tejido tumoral— mostraron una concordancia mucho mayor entre TempO-Seq y HTG. En la mayoría de los casos, las puntuaciones o clasificaciones fueron similares, incluso después de que los investigadores simularan el uso de menos lecturas de secuenciación para imitar diferentes capacidades de máquina.
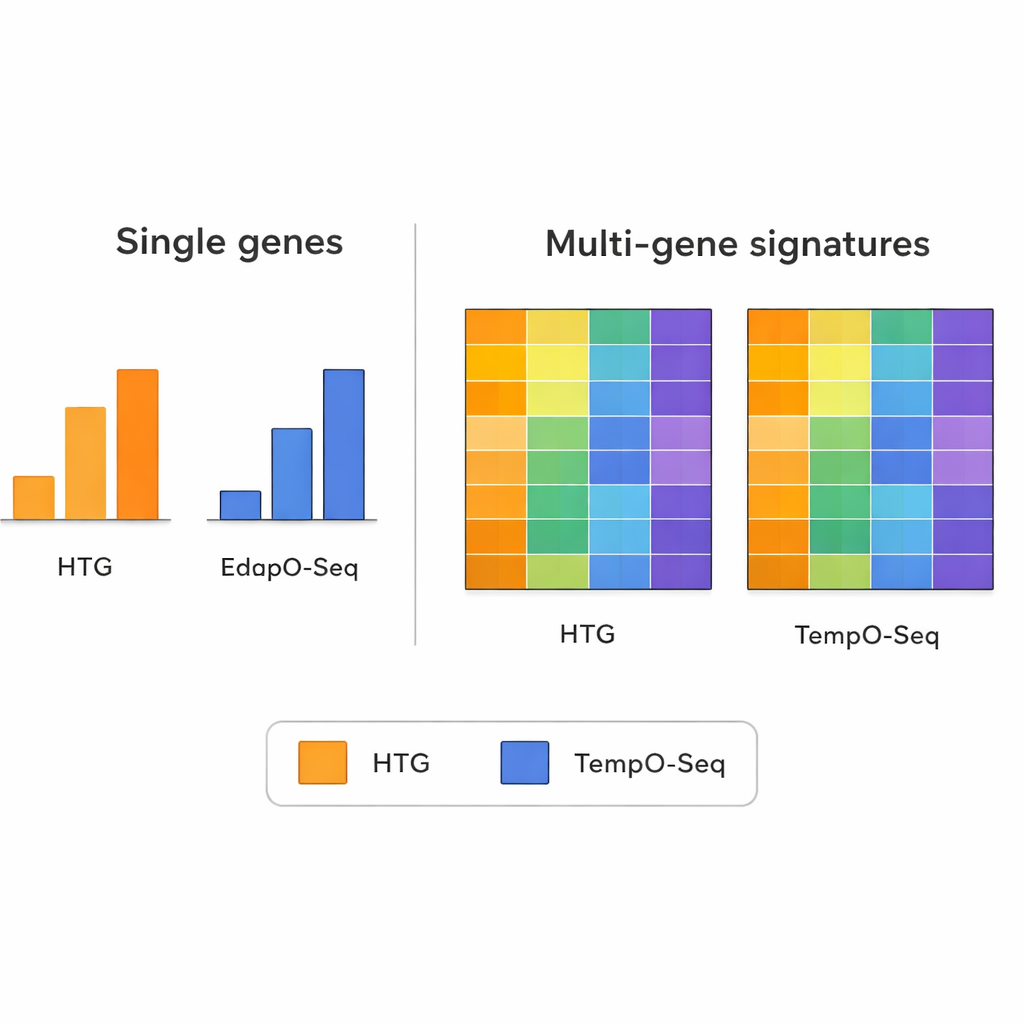
Patrones multigénicos como señales fiables
El estudio subraya un principio importante en la genómica moderna: mientras que la medición de un único gen puede verse afectada por peculiaridades técnicas, combinar señales de docenas o cientos de genes tiende a promediar ese ruido. Los autores usaron varias herramientas multigénicas bien conocidas como pruebas de estrés técnico. Estas incluyeron un panel de cáncer de mama que asigna tumores a subtipos intrínsecos, un algoritmo que puntúa cuánto tejido inmune y conectivo está mezclado en una muestra tumoral, y un método que estima las proporciones de muchos tipos de células inmunitarias. En estos análisis complejos, TempO-Seq suele seguir de cerca a HTG, lo que sugiere que captura las mismas historias biológicas aun cuando algunos detalles finos difieran.
Qué implica esto de cara al futuro
Para los investigadores que dependen de los archivos FFPE para estudiar el cáncer, la pérdida de una plataforma de confianza podría haber sido un revés importante. Este estudio de referencia ofrece tranquilidad: TempO-Seq parece ser un sustituto sólido de HTG HTP cuando el objetivo es usar biomarcadores multigénicos y patrones de expresión amplios, que son la columna vertebral de muchas herramientas diagnósticas y pronósticas modernas. Los autores advierten que comparar directamente resultados de genes individuales entre plataformas no es aconsejable, porque cada método dirige los genes de maneras ligeramente distintas. En su lugar, recomiendan centrarse en firmas complejas y multigénicas para trabajos entre plataformas. En términos sencillos, el nuevo método parece capaz de continuar la labor de su predecesor para la mayoría de las necesidades reales de la investigación oncológica, especialmente cuando a los científicos les importa el patrón global de muchos genes más que el valor exacto de un solo gen.
Cita: Fernández-Serra, A., López-Reig, R., Romero, I. et al. Comparative evaluation of HTG and TempO Seq targeted transcriptome profiling methods. Sci Rep 16, 6108 (2026). https://doi.org/10.1038/s41598-026-36810-w
Palabras clave: perfilado transcriptómico, cáncer de endometrio, tejido FFPE, secuenciación dirigida de ARN, biomarcadores de expresión génica