Clear Sky Science · es
Un enfoque de aprendizaje automático para predecir coeficientes osmóticos y derivar coeficientes de actividad en sales de amonio alquilo
Productos cotidianos con complejidad oculta
Desde suavizantes y acondicionadores hasta toallitas desinfectantes y enjuagues bucales, una familia de compuestos llamada sales de amonio cuaternario—a menudo abreviada como “Quat”—impulsa discretamente muchos productos que usamos. Ayudan a eliminar gérmenes, ablandar la ropa y acelerar reacciones industriales. Sin embargo, predecir con exactitud cómo se comportan estas sales en agua ha sido sorprendentemente difícil, lo que limita la eficiencia con que podemos diseñar formulaciones más seguras y sostenibles. Este estudio muestra cómo el aprendizaje automático moderno puede aprender de mediciones previas para predecir ese comportamiento de forma más flexible y, en muchos casos, con mayor precisión que los modelos tradicionales.
Por qué importan estas sales
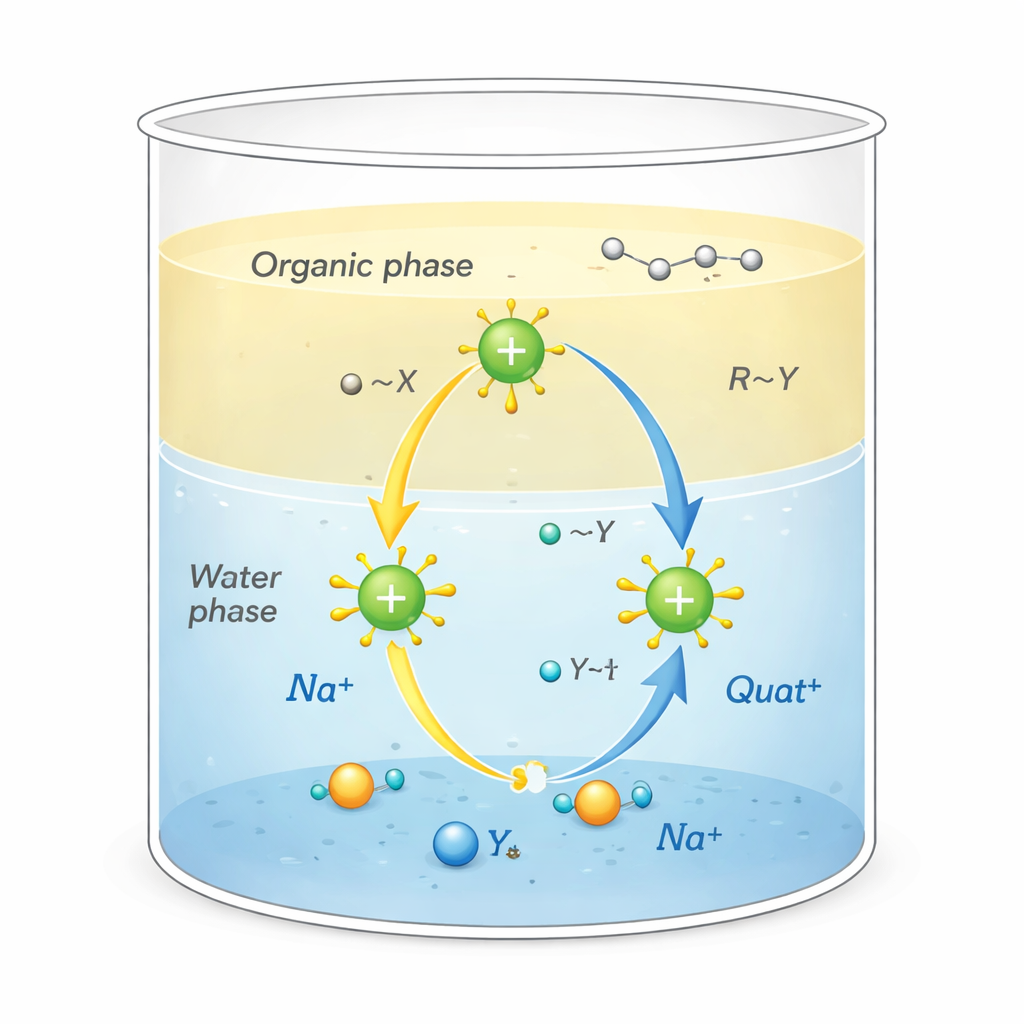
Los Quats son moléculas con carga positiva rodeadas por colas ricas en carbono. Esta forma inusual les permite cumplir varias funciones a la vez: adherirse a suciedad oleosa, fijarse a superficies como tejidos o cabello y alterar las membranas de los microbios, lo que las convierte en potentes desinfectantes y tensioactivos. También se usan como catalizadores por transferencia de fase, actuando como transportadores que llevan iones reactivos desde el agua hacia disolventes de tipo oleoso donde normalmente no irían. Esa acción de traslado, que ocurre en la interfase entre agua y aceite, puede acelerar de forma notable reacciones químicas empleadas en la fabricación de fármacos, polímeros y productos químicos finos.
Por qué es difícil predecir su comportamiento
Para diseñar nuevos Quats o ajustar los existentes, los químicos necesitan saber cómo se comportan en solución—qué tan fuertemente interactúan con el agua y con otros iones disueltos. Dos medidas clave son el coeficiente osmótico, que refleja cómo las sales afectan la tendencia del agua a atravesar membranas, y el coeficiente de actividad, que captura cuán “efectiva” es una especie disuelta en comparación con una solución ideal perfectamente mezclada. Tradicionalmente, estos valores se obtienen mediante experimentos laboriosos o usando modelos físicos complejos como Electrolyte‑NRTL y Extended UNIQUAC, que requieren muchos parámetros ajustados y no son fáciles de generalizar a moléculas nuevas.
Enseñar a un ordenador a leer moléculas
Los investigadores tomaron una ruta distinta: preguntaron si un ordenador podía aprender la relación entre la estructura de los Quats y su comportamiento osmótico directamente a partir de datos existentes. Reunieron 1.654 mediciones de coeficientes osmóticos para 52 Quats diferentes extraídas de la literatura científica. Cada molécula se describió usando la notación SMILES—una representación en cadena que codifica rasgos como el número de átomos de carbono y oxígeno, la presencia de anillos de benceno, ramificaciones y el tipo de grupo de nitrógeno con carga positiva, junto con el ion negativo acompañante (como cloruro, bromuro o nitrato). Estos descriptores estructurales, además de la concentración de la sal, sirvieron como entradas para varios algoritmos de aprendizaje supervisado implementados en Python.
Encontrar el predictor más fiable
Siete algoritmos diferentes, incluyendo regresión lineal, árboles de decisión, bosques aleatorios, máquinas de vectores de soporte, gradient boosting, k‑vecinos más cercanos y procesos gaussianos, se entrenaron con el 70% de los datos y se probaron con el 30% restante. El equipo también usó un esquema de validación más estricto en el que se dejaban fuera todos los datos de una sal para ver qué tan bien los modelos extrapolaban a un compuesto realmente no visto. La regresión lineal funcionó mal, perdiéndose tendencias no lineales importantes. Los métodos basados en árboles ajustaron muy bien los datos de entrenamiento pero produjeron predicciones algo irregulares y perdieron precisión con sales nuevas. El modelo de proceso gaussiano logró el mejor equilibrio: entregó curvas suaves y físicamente razonables para los coeficientes osmóticos y consiguió un error porcentual absoluto medio de alrededor del 5% en general, superando a otros enfoques de aprendizaje automático en las pruebas más exigentes.
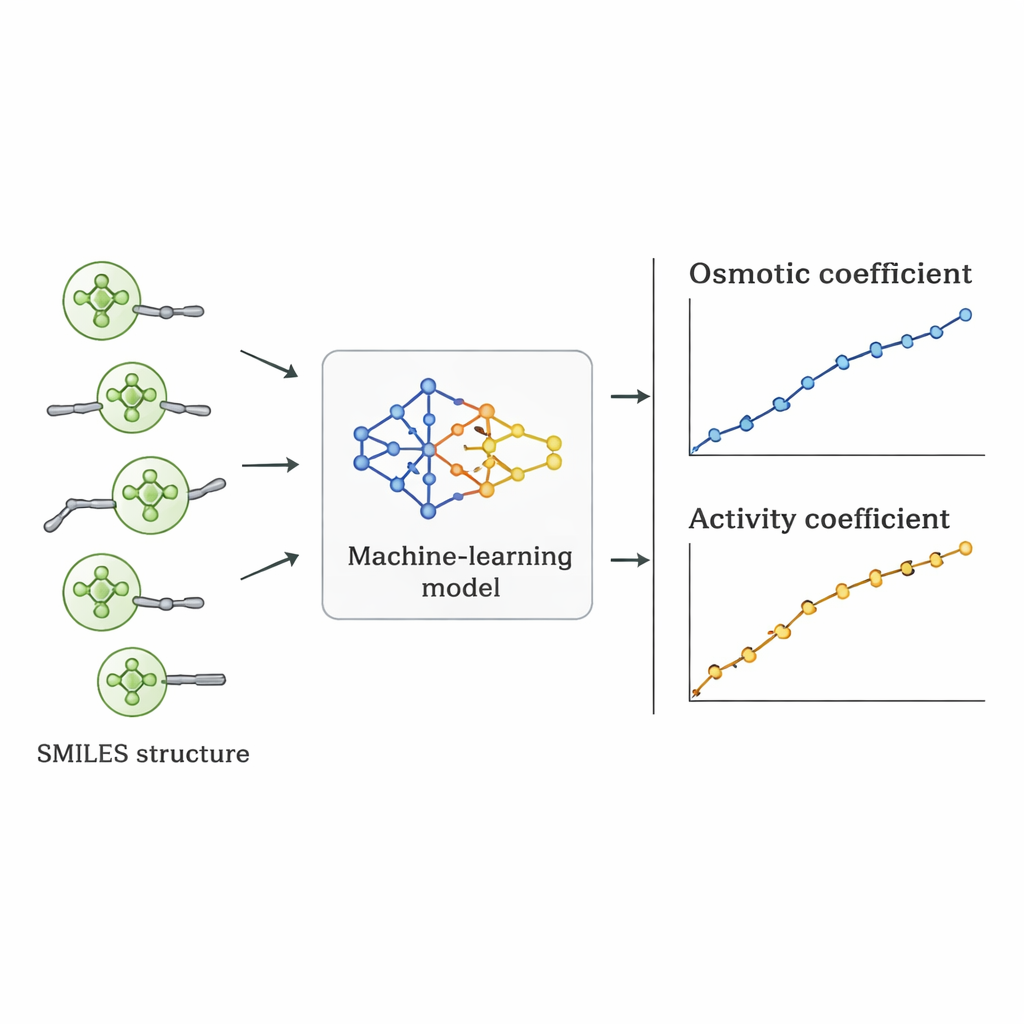
Del comportamiento osmótico a números útiles para el diseño
Una vez elegido el mejor modelo, sus coeficientes osmóticos predichos se convirtieron en coeficientes de actividad usando relaciones termodinámicas estándar. Cuando estos coeficientes de actividad se compararon con valores derivados de experimentos y de modelos físicos establecidos, el enfoque de aprendizaje automático con frecuencia los igualó o los superó para Quats individuales. Aunque su error medio en todas las sustancias fue ligeramente mayor que el de algunos modelos especializados, tenía una ventaja crucial: al estar impulsado por descriptores estructurales en lugar de ajustes específicos por sal, puede aplicarse a Quats nuevos que nunca se han medido en el laboratorio, siempre que sus estructuras se parezcan a las del conjunto de entrenamiento.
Qué significa esto para productos y procesos
Para un público no especializado, el mensaje es que los ordenadores ahora pueden “leer” descripciones compactas en texto de moléculas y, a partir de patrones aprendidos en datos previos, predecir con notable precisión cómo se comportarán en agua. Esto abre la puerta a un cribado más rápido y barato de nuevos Quats para desinfectantes, limpiadores, productos de cuidado personal y catalizadores industriales, sin experimentación exhaustiva para cada candidato. El modelo actual es solo un primer paso, y los autores señalan que huellas moleculares más ricas y algoritmos más recientes podrían mejorar aún más el rendimiento. Aun así, demuestra cómo las herramientas basadas en datos pueden complementar la química tradicional, ayudando a los ingenieros a diseñar formulaciones más eficaces y potencialmente más seguras al explorar posibilidades químicas que sería impráctico probar una por una en el laboratorio.
Cita: Chawuthai, R., Murathathunyaluk, S., Saengsuradech, S. et al. A machine learning approach for predicting osmotic coefficients and deriving activity coefficients in alkyl ammonium salts. Sci Rep 16, 5969 (2026). https://doi.org/10.1038/s41598-026-36758-x
Palabras clave: sales de amonio cuaternario, catálisis por transferencia de fase, coeficientes osmóticos, coeficientes de actividad, aprendizaje automático en química