Clear Sky Science · es
Marco híbrido de aprendizaje profundo para la clasificación precisa de datos genómicos de alta dimensión
Entendiendo la avalancha de datos genómicos
Las tecnologías modernas de ADN pueden medir decenas de miles de genes en un solo experimento, prometiendo detección temprana de enfermedades y tratamientos más precisos. Sin embargo, esta abundancia de datos es tan grande, ruidosa y compleja que incluso modelos informáticos potentes suelen tener dificultades para encontrar patrones claros y fiables. Este artículo presenta un nuevo tipo de sistema de inteligencia artificial (IA) diseñado específicamente para manejar esos datos genómicos abrumadores, con el objetivo de hacer las predicciones más precisas y, al mismo tiempo, explicar cómo se han tomado dichas decisiones.
Por qué los datos genómicos son tan difíciles de usar
Los estudios genómicos producen rutinariamente muchas más mediciones que pacientes o muestras. Muchas de estas mediciones son irrelevantes, redundantes o están distorsionadas por ruido técnico. Los métodos tradicionales de aprendizaje automático requieren que expertos humanos seleccionen manualmente qué genes pueden importar, o intentan usar todo y corren el riesgo de sobreajustar: desempeñarse bien en los datos de entrenamiento pero fallar en casos nuevos. El aprendizaje profundo, que ha transformado campos como el reconocimiento de imágenes, puede aprender patrones automáticamente a partir de datos crudos. Sin embargo, en genómica suele comportarse como una caja negra: puede ofrecer respuestas precisas, pero da poca información sobre por qué, lo que limita su aceptación en medicina, donde la transparencia es esencial.
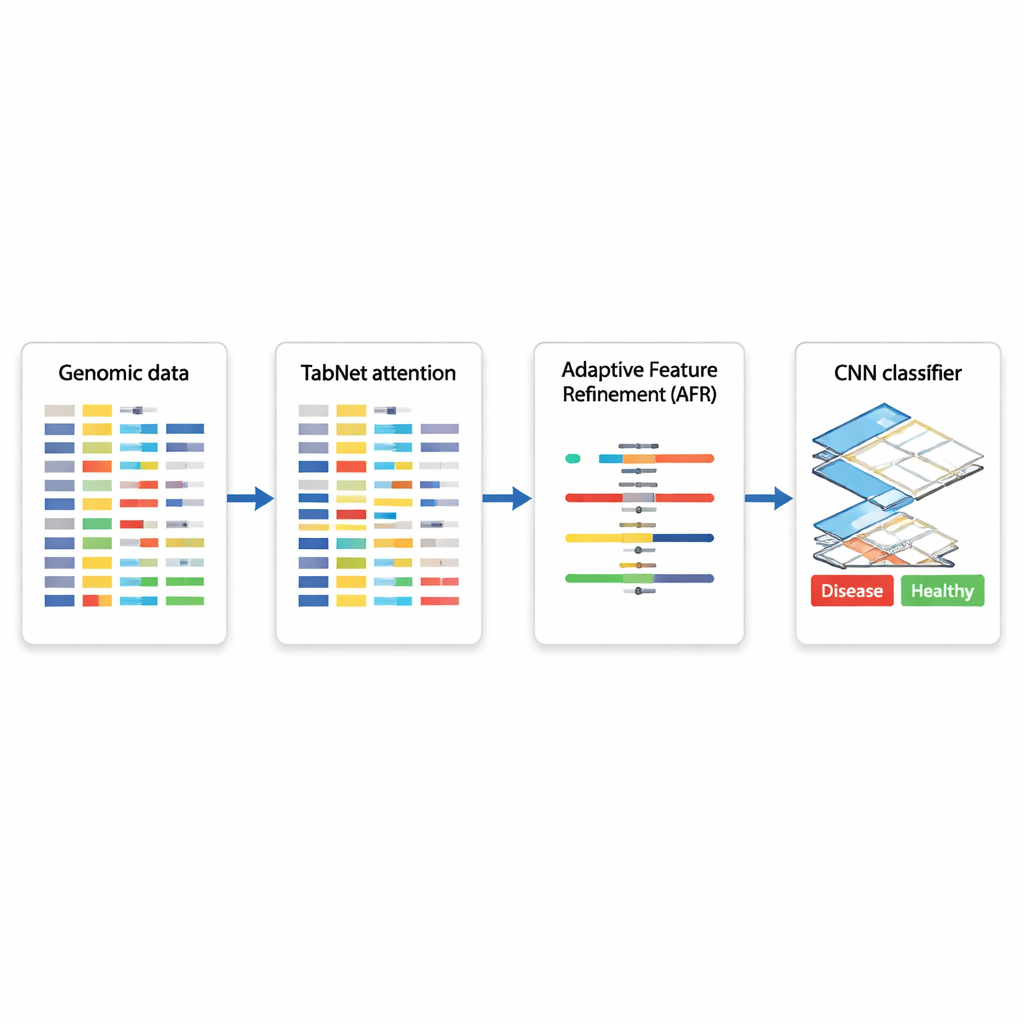
Un plano híbrido de IA para decisiones basadas en genes
Los autores proponen una arquitectura híbrida de aprendizaje profundo que encadena tres módulos especializados. Primero, un componente llamado TabNet actúa como un foco, escaneando todas las mediciones genómicas disponibles y aprendiendo qué características son más informativas para una tarea dada —por ejemplo, distinguir tejido canceroso de tejido no canceroso. En lugar de tratar cada gen por igual, TabNet centra la atención en un subconjunto escaso que parece más relevante. A continuación, una capa de Refinamiento Adaptativo de Características (AFR) toma estas señales seleccionadas y las repondera, reforzando patrones consistentes y significativos mientras atenúa aún más el ruido. Finalmente, una red neuronal convolucional (CNN), comúnmente usada en análisis de imágenes, examina cómo interactúan localmente las características refinadas, capturando relaciones sutiles entre grupos de genes que podrían indicar un subtipo de enfermedad o un estado biológico particular.
Poniendo el modelo a prueba
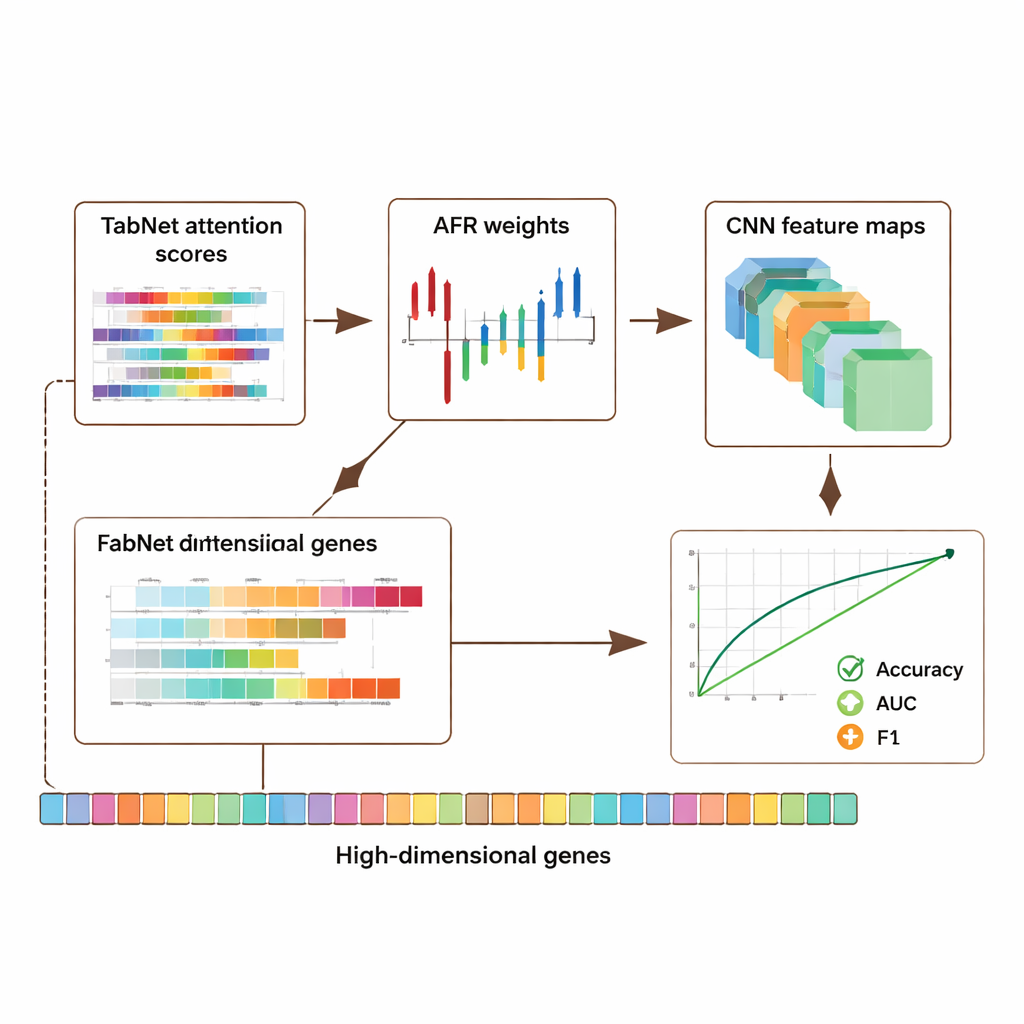
El marco se evaluó en tres recursos públicos principales: un conjunto de datos de cáncer de mama del The Cancer Genome Atlas, un conjunto de datos de melanoma de células individuales del Gene Expression Omnibus y un conjunto epigenómico del proyecto ENCODE. En conjunto, estas colecciones incluyen miles de muestras y decenas de miles de características por muestra, cubriendo actividad génica y marcas químicas sobre el ADN. En todos los conjuntos, el modelo híbrido superó a varios enfoques de vanguardia, mejorando la precisión y medidas clave de calidad de clasificación como el área bajo la curva ROC (AUC) y la puntuación F1 en aproximadamente 5–8 puntos porcentuales. Es importante destacar que estas mejoras no comprometieron la transparencia: el modelo produce mapas de atención de TabNet y mapas de activación de la CNN que resaltan qué genes y regiones fueron más influyentes en cada predicción.
Equilibrando precisión, privacidad y confianza
Dado que los datos genómicos son profundamente personales, los autores también estudiaron cómo proteger la privacidad conservando la señal útil. Introdujeron un mecanismo de privacidad adaptativo que añade más ruido a características altamente sensibles y menos a otras, combinado con enmascaramiento de entradas seleccionadas. Las pruebas mostraron que incluso cuando se introdujo ruido moderado, el modelo mantuvo una fuerte precisión y capacidad de discriminación, degradándose de forma gradual a medida que se aumentaba la protección. Al mismo tiempo, los patrones interpretables de atención y activación señalaron con frecuencia genes ya conocidos por su papel en el cáncer y la regulación inmune, lo que sugiere que el sistema no se limita a memorizar datos sino que captura señales biológicamente significativas. Un estudio de ablación —eliminando sistemáticamente partes de la arquitectura— confirmó que cada módulo, especialmente la capa AFR, aportó de forma medible al rendimiento.
Qué significa esto para la medicina del futuro
En términos sencillos, este trabajo ofrece una forma más inteligente de cribar enormes hojas de cálculo genómicas para encontrar patrones vinculados a la enfermedad, al tiempo que muestra qué entradas de la hoja fueron las más relevantes. Al combinar selección dirigida de características, refinamiento cuidadoso y reconocimiento de patrones, el modelo híbrido mejora la precisión de las predicciones, sigue siendo gestionable computacionalmente y proporciona pistas visuales que los clínicos y biólogos pueden interpretar. Aunque se necesitan más pruebas en grupos de pacientes más amplios y diversos, estos marcos podrían ayudar a identificar nuevos biomarcadores, refinar subtipos de enfermedad y apoyar herramientas de decisión clínica en medicina de precisión —acercando el análisis de ADN con IA un paso más hacia el uso real en el mundo clínico.
Cita: Swain, M.K., Kamila, N.K., Jena, L. et al. Hybrid deep learning framework for accurate classification of high dimensional genomic data. Sci Rep 16, 5919 (2026). https://doi.org/10.1038/s41598-026-36128-7
Palabras clave: aprendizaje profundo genómico, descubrimiento de biomarcadores del cáncer, IA interpretable, medicina de precisión, genómica con preservación de la privacidad