Clear Sky Science · es
Un estudio cuantitativo de compuestos citotóxicos usando descriptores basados en grafos y aprendizaje automático
Por qué esta investigación importa para futuros fármacos contra el cáncer
Los fármacos contra el cáncer que matan células tumorales, conocidos como fármacos citotóxicos, a menudo caminan por la cuerda floja entre salvar vidas y causar efectos secundarios graves. Para diseñar tratamientos más seguros y efectivos, los científicos necesitan métodos rápidos y fiables para predecir cómo se desplazan estos fármacos por el cuerpo: cuán bien se absorben, con qué facilidad atraviesan las membranas celulares y dónde terminan. Este estudio muestra cómo descripciones matemáticas de las moléculas de los fármacos, combinadas con aprendizaje automático moderno, pueden estimar con precisión una propiedad clave que controla este comportamiento, acelerando potencialmente la búsqueda de mejores terapias contra el cáncer.
Una superficie clave que controla adónde pueden ir los fármacos
Una idea central en el artículo es el área polar superficial topológica, o Top_PSA. En términos sencillos, este es un número que refleja cuánto de la superficie de una molécula está constituida por regiones “polares”: partes que gustan del agua y que pueden formar enlaces de hidrógeno. Las moléculas con áreas polares superficiales muy altas suelen tener dificultades para atravesar membranas grasas y pueden absorberse mal por vía oral. Las moléculas con áreas polares superficiales muy bajas pueden cruzar demasiadas barreras con facilidad, a veces causando efectos indeseados en tejidos sensibles como el cerebro. Top_PSA se ha convertido en un atajo popular para estimar estas propiedades de transporte porque puede calcularse rápidamente a partir de un dibujo 2D de la molécula, sin necesidad de simulaciones 3D lentas.
Convertir dibujos moleculares en números
Los investigadores reunieron un conjunto curado de 156 compuestos citotóxicos diferentes procedentes de fármacos anticancerígenos reales y de agentes experimentales. Luego convirtieron cada molécula en 58 denominados descriptores: números que capturan características como cuántos átomos tiene, cuántos anillos, cuán flexibles son sus enlaces, cuántos átomos pueden formar enlaces de hidrógeno y cuán polares o electronegativos son diferentes fragmentos. Muchos de estos descriptores provienen de la teoría de grafos, donde una molécula se trata como una red de nodos y enlaces conectados. Este rico retrato numérico de cada molécula sirvió como entrada para modelos computacionales que intentaron predecir los valores de Top_PSA calculados por conjuntos de herramientas químicas ampliamente utilizados.
Probar múltiples caminos hacia una predicción precisa
Para encontrar la mejor manera de vincular estos descriptores con Top_PSA, el equipo comparó varias estrategias de modelado. Probaron regresión lineal estándar así como dos versiones “regularizadas” llamadas ridge y regresión LASSO, diseñadas para manejar mejor información ruidosa y solapada. También exploraron diferentes esquemas de preparación de datos: ajustar modelos directamente a los descriptores sin procesar, comprimirlos con análisis de componentes principales (PCA), escalarlos de una forma que reduzca el impacto de valores extremos (escalado robusto), ajustar valores atípicos y podar características altamente correladas usando una medida llamada factor de inflación de la varianza. Cada enfoque se evaluó cuidadosamente usando validación cruzada k‑fold, un método que divide repetidamente los datos en subconjuntos de entrenamiento y prueba para proteger contra el sobreajuste.
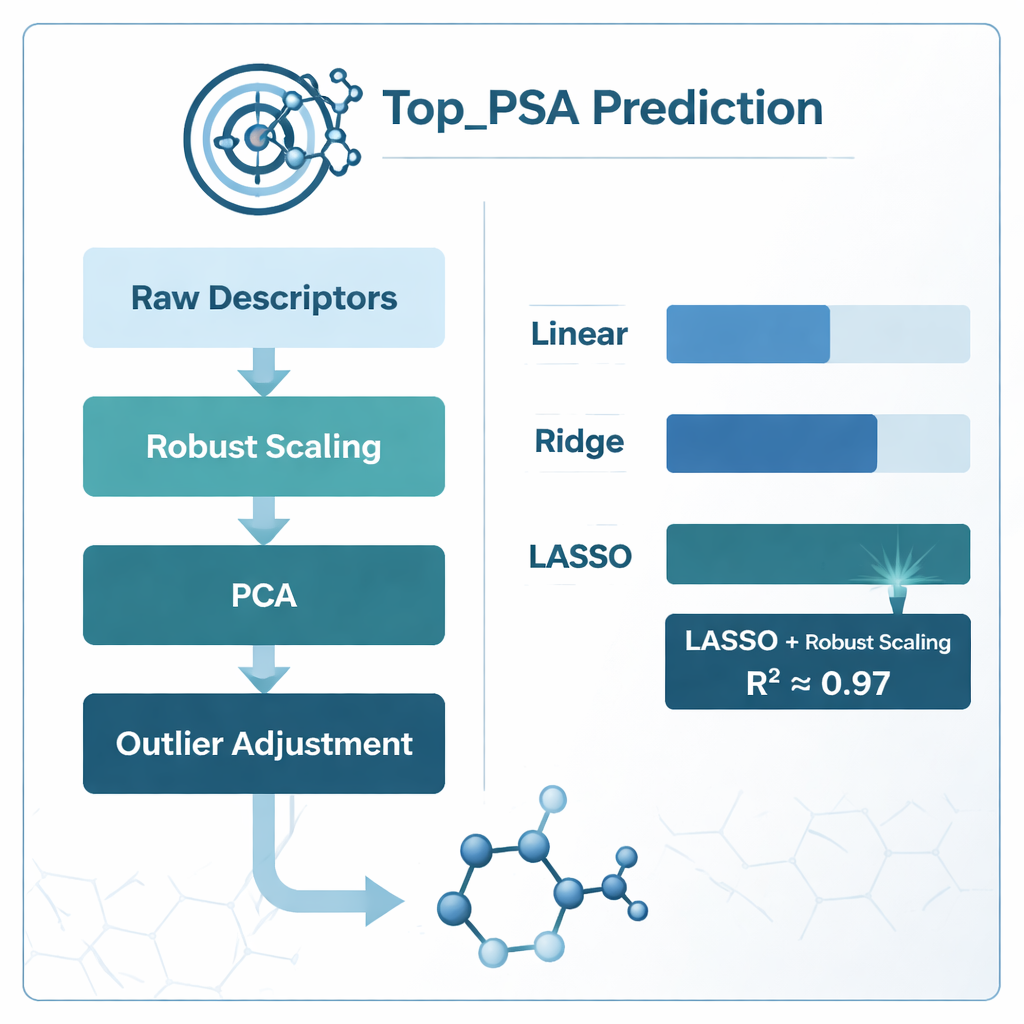
Qué funcionó mejor y qué aprendieron los modelos
El claro ganador fue la combinación de escalado robusto con regresión LASSO, que alcanzó un coeficiente de determinación (R²) de aproximadamente 0,97 —lo que significa que pudo explicar alrededor del 97 % de la variación en Top_PSA entre los 156 fármacos. Los modelos basados en PCA se acercaron en precisión bruta pero fueron más difíciles de interpretar químicamente porque los descriptores originales se mezclan en componentes abstractos. La simple poda de descriptores correlacionados usando el factor de inflación de la varianza en realidad perjudicó el rendimiento, lo que sugiere que algunas medidas solapadas todavía contienen información química útil. Al examinar qué pesos de descriptores LASSO mantuvo diferentes de cero, los autores encontraron que los factores más importantes eran la presencia de heteroátomos como nitrógeno y oxígeno, la capacidad de donar o aceptar enlaces de hidrógeno y los índices que siguen cómo se disponen los átomos electronegativos en el grafo molecular —todas características que coinciden con la comprensión química intuitiva del área polar superficial.
Cómo esto puede guiar un mejor diseño de fármacos
Para lectores fuera del campo, el mensaje clave es que huellas matemáticas de las moléculas preparadas con cuidado, cuando se emparejan con métodos de aprendizaje automático bien elegidos, pueden proporcionar estimaciones rápidas y fiables de cuán “pegajosos” o “resbaladizos” serán los fármacos contra el cáncer a medida que viajan por el cuerpo. El estudio ofrece orientación práctica para otros investigadores sobre cómo preprocesar datos de descriptores, qué enfoques de modelado favorecer y qué atajos evitar. A la larga, modelos robustos e interpretables de Top_PSA pueden ayudar a los químicos a filtrar enormes bibliotecas virtuales de posibles fármacos, centrando sus esfuerzos en compuestos con el equilibrio adecuado entre cruce de membrana y seguridad —un paso importante hacia tratamientos contra el cáncer más efectivos y menos tóxicos.
Cita: Ahmad, S., Javed, S., Khalid, S. et al. A quantitative study of cytotoxic compounds using graph based descriptors and machine learning. Sci Rep 16, 5076 (2026). https://doi.org/10.1038/s41598-026-35728-7
Palabras clave: fármacos citotóxicos, superficie polar, descriptores moleculares, aprendizaje automático, permeabilidad de fármacos