Clear Sky Science · es
La disfunción mitocondrial y la desregulación de Ca2+ en neuronas humanas derivadas de iPSC portadoras de la mutación presenilina‑1 surgen bajo estrés mediante un mecanismo independiente de MCU‑1
Por qué esto importa para la enfermedad de Alzheimer
La enfermedad de Alzheimer suele describirse en términos de placas proteicas pegajosas en el cerebro, pero mucho antes de que la memoria empiece a fallar, las pequeñas “centrales energéticas” dentro de las neuronas —las mitocondrias— y el manejo de los iones calcio pueden estar ya desajustándose. Este estudio utiliza neuronas humanas cultivadas a partir de células de la piel de una persona portadora de una conocida mutación familiar de Alzheimer para plantear una pregunta simple pero crucial: ¿qué tan pronto, y de qué manera, empiezan a fallar la producción de energía y el equilibrio de calcio?
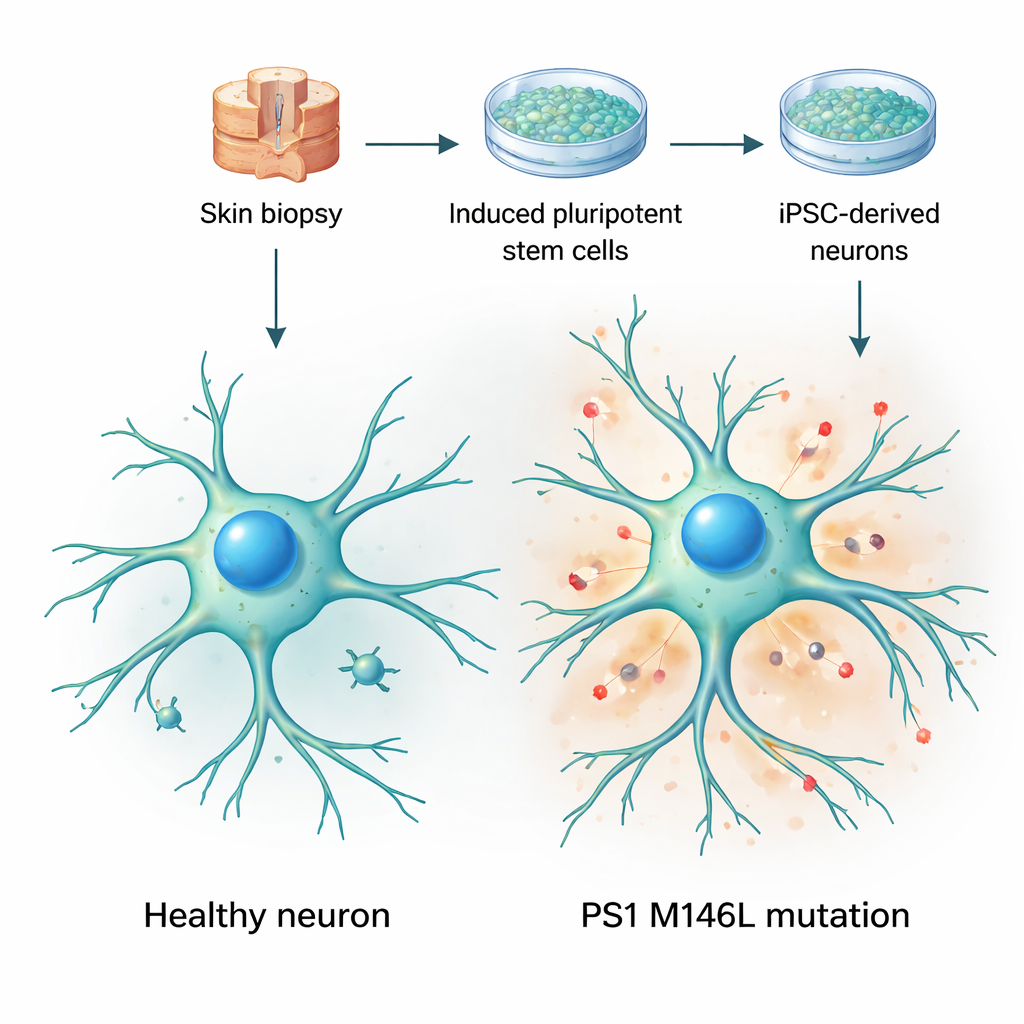
Convertir células de la piel en modelos cerebrales vivos
Los investigadores partieron de biopsias de piel de dos mujeres: una voluntaria sana y una portadora asintomática de la mutación de presenilina‑1 denominada M146L, presente en una familia argentina con Alzheimer de inicio temprano. Reprogramaron las células de la piel a células madre pluripotentes inducidas —células que pueden convertirse en casi cualquier tejido— y luego las dirigieron para que se diferenciaran en neuronas. Durante varias semanas en cultivo, estas células adquirieron formas neuronales típicas, extendieron procesos largos y ramificados y expresaron marcadores neuronales habituales. Es importante que tanto las células control como las mutantes maduraron a ritmos similares y parecían en general saludables, lo que permitió al equipo centrarse en cambios funcionales sutiles en lugar de en una pérdida celular evidente o daños claros.
Señales eléctricas y calcio bajo tensión
Las neuronas dependen de un control estricto del calcio, un átomo cargado que actúa como un interruptor rápido para muchos procesos celulares. Usando tintes fluorescentes, el equipo registró cómo variaban los niveles de calcio dentro de las células cuando eran estimuladas eléctricamente con potasio o activadas con moléculas señalizadoras. Bajo una estimulación despolarizante simple, las neuronas portadoras de la mutación M146L mostraron aumentos de calcio más débiles que las neuronas control, lo que sugiere problemas para mantener los gradientes eléctricos e iónicos que normalmente impulsan la entrada de calcio. Sin embargo, cuando los investigadores provocaron una situación más estresante —forzando la liberación de calcio desde los depósitos internos en el retículo endoplásmico— la diferencia se hizo más evidente. En respuesta a este estrés, las mitocondrias de las neuronas mutantes tomaron significativamente menos calcio que las de las células control, lo que indica una capacidad reducida para amortiguar picos peligrosos de calcio.
Desacoplando el uso de energía del equilibrio del calcio
Para entender cómo este manejo alterado del calcio afecta el metabolismo celular, los investigadores midieron cuánto oxígeno consumían las neuronas —un indicador directo de la actividad mitocondrial. Sorprendentemente, las neuronas con la mutación M146L respiraban más: sus tasas de consumo de oxígeno basal y máxima, así como la cantidad de oxígeno vinculada a la producción de ATP, fueron todas más altas que en las células control. Aun así, la eficiencia en el acoplamiento del uso de oxígeno a la producción de ATP parecía similar, y no hubo un aumento en el número de mitocondrias ni en enzimas clave para fabricar ATP. En cambio, las mitocondrias en las neuronas mutantes eran más largas y tubulares, con niveles más altos de una proteína de fusión llamada mitofusina‑1, un patrón que suele observarse en células sometidas a estrés crónico y de bajo nivel. Estas mitocondrias hiperactivas y alargadas también generaron más especies reactivas de oxígeno, moléculas inestables que pueden dañar proteínas y ADN si no se controlan adecuadamente.
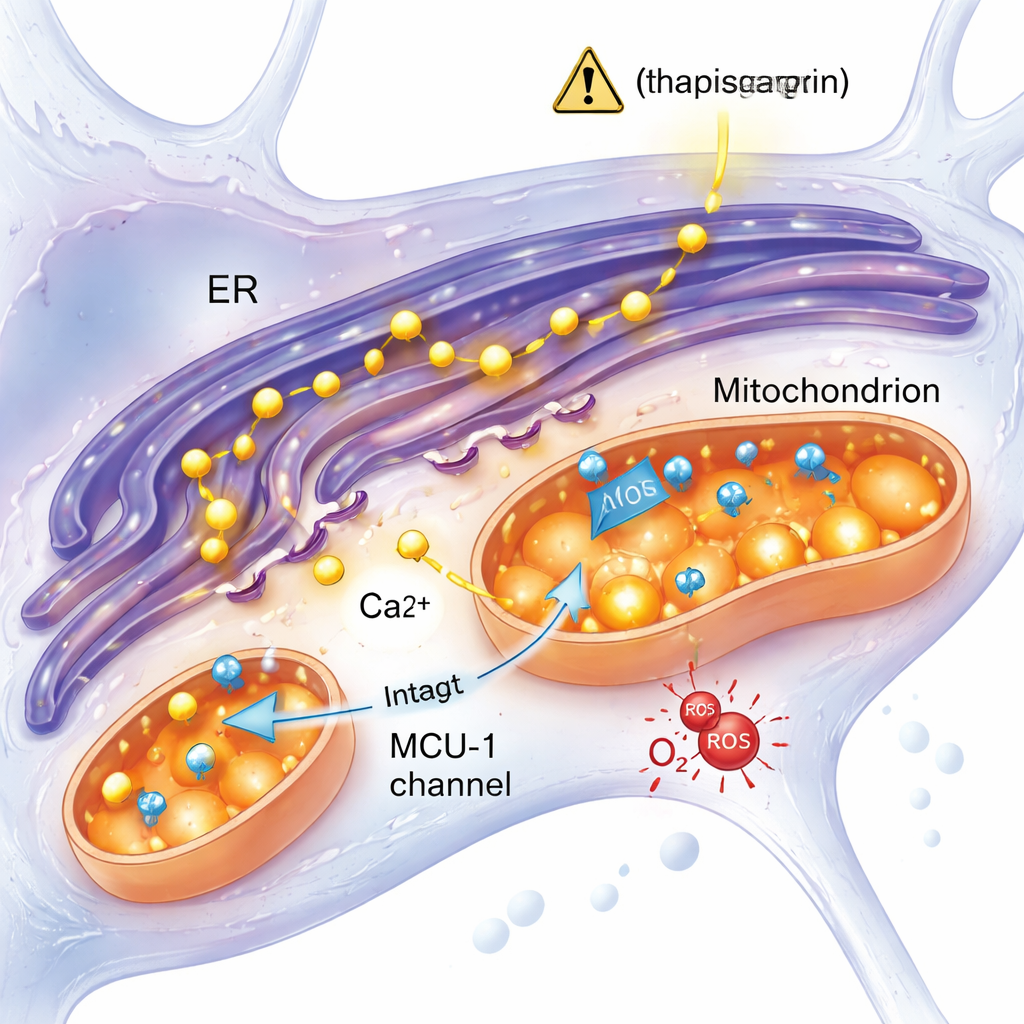
Una respuesta al estrés independiente de un canal clave de calcio
Una idea principal en la investigación del Alzheimer es que un exceso de calcio del retículo endoplásmico fluye hacia las mitocondrias a través de un canal llamado uniportador de calcio mitocondrial (MCU‑1), sobrecargándolas y provocando disfunción. Este estudio puso a prueba esa noción directamente. Cuando el equipo bloqueó MCU‑1 con un inhibidor específico, tanto las neuronas control como las mutantes mostraron fuertes reducciones en la captación mitocondrial de calcio, confirmando que el canal en sí funcionaba en ambos grupos. Además, cuando la liberación de calcio se desencadenó a través de una vía más fisiológica que implicaba al receptor IP3 —otra puerta clave del calcio— las células mutantes y control respondieron de forma similar. Estos resultados alejan la hipótesis de un MCU‑1 defectuoso y, en su lugar, sugieren que los contactos físicos y funcionales entre el retículo endoplásmico y las mitocondrias, u otros aspectos de su interacción, están alterados en las neuronas mutantes.
Qué significa esto para entender y tratar la enfermedad
En conjunto, los hallazgos dibujan el retrato de neuronas humanas portadoras de la mutación PS1 M146L como células que parecen normales en reposo pero reaccionan de forma anómala bajo estrés. Sus mitocondrias no captan suficiente calcio cuando los depósitos internos se liberan de repente, y, sin embargo, funcionan más intensamente —consumiendo más oxígeno y generando más especies reactivas de oxígeno— como si estuvieran atrapadas en un modo compensatorio costoso. Dado que esto ocurre en neuronas derivadas de humanos vivos antes de cualquier síntoma clínico, el trabajo respalda la idea de que la alteración de la señalización del calcio y el sobretrabajo mitocondrial temprano son eventos aguas arriba en el Alzheimer, no solo subproductos tardíos. Para los no especialistas, el mensaje clave es que mantener el equilibrio entre las señales de calcio y la producción de energía mitocondrial puede ser tan central para prevenir la enfermedad como atacar las placas de amiloide más conocidas.
Cita: Wilson, C., Galeano, P., Remedi, M.M. et al. Mitochondrial dysfunction and Ca2+ dysregulation in human iPSC-derived neurons carrying presenilin-1 mutation arise under stress via an MCU-1-independent mechanism. Sci Rep 16, 6002 (2026). https://doi.org/10.1038/s41598-026-35597-0
Palabras clave: Enfermedad de Alzheimer, mitocondrias, señalización del calcio, mutación de presenilina‑1, neuronas derivadas de iPSC