Clear Sky Science · es
Evaluación de la base bioquímica que subyace a la resistencia frente a la amiloidosis sistémica
Cuando pequeños cambios proteicos bloquean una acumulación letal
Muchas enfermedades inflamatorias crónicas, desde la artritis reumatoide hasta la tuberculosis, pueden desencadenar una complicación rara pero a menudo fatal llamada amiloidosis sistémica AA. En esta condición, una proteína normal de la sangre se acumula en forma de fibras rígidas que obstruyen órganos. Este estudio plantea una pregunta sorprendentemente esperanzadora: ¿pueden pequeños cambios naturales en esa proteína hacer que algunos animales sean en gran medida inmunes a la enfermedad y, de ser así, cómo?
La amenaza oculta de las acumulaciones proteicas
La amiloidosis AA comienza con una señal inflamatoria en la sangre llamada amiloide sérico A (SAA). Durante inflamaciones intensas o prolongadas, los niveles de SAA pueden aumentar miles de veces por encima de lo normal. En algunas personas y animales, una porción de esta proteína se pliega mal y se apila en fibras largas, conocidas como fibrillas amiloides, que se extienden por órganos como el bazo y los riñones. Con el tiempo, esas fibras comprometen la función de los órganos. Sin embargo, no todos los individuos con niveles altos de SAA desarrollan amiloidosis, y ciertas cepas de ratón resultan sorprendentemente resistentes incluso cuando en el laboratorio se las empuja hacia la enfermedad. Entender por qué podría señalar nuevas estrategias para prevenir la acumulación de amiloide en humanos.
Ratones resistentes y sus versiones proteicas especiales
En ratones, la mayoría de las fibrillas amiloides AA se originan a partir de una versión de SAA denominada SAA1.1, que está fuertemente ligada a la enfermedad. Sin embargo, algunas cepas de ratón producen principalmente versiones ligeramente alteradas, llamadas SAA1.5 y SAA2.2, y estas cepas rara vez desarrollan amiloidosis sistémica AA. Las proteínas difieren solo en un puñado de bloques constructivos (aminoácidos), pero esos cambios se agrupan en una región compacta que forma el núcleo interno de las fibras causantes de la enfermedad. Los investigadores propusieron que estas pequeñas diferencias no impiden que las proteínas se agreguen por completo, sino que les impiden adoptar la forma fibrilar muy específica que resulta dañina.
Poniendo a prueba las proteínas en el laboratorio
Para explorar esta idea, el equipo produjo las tres variantes de SAA de ratón en bacterias y observó su comportamiento en experimentos de tubo de ensayo. Monitorearon la formación de fibrillas usando un tinte fluorescente que se ilumina cuando se forma amiloide y verificaron las estructuras con microscopía electrónica. La SAA1.1 vinculada a la enfermedad formó con facilidad fibrillas largas y rectas. La SAA2.2 también pudo formar fibrillas, pero eran más gruesas, con más torsión y mayor diversidad estructural, y no generaron la misma señal fuerte del tinte. En contraste, la SAA1.5 no logró formar fibrillas bajo las condiciones probadas. Cuando los científicos añadieron pequeñas muestras de fibrillas reales tomadas del bazo de ratones enfermos como “semillas”, la SAA1.1 creció rápidamente formando nuevas fibrillas que copiaban de cerca la estructura del original, tal como ocurre con un prion. De forma llamativa, la SAA1.5 y la SAA2.2 no se incorporaron a estas semillas en absoluto; las fibras ex vivo no pudieron reclutarlas hacia la forma patógena.
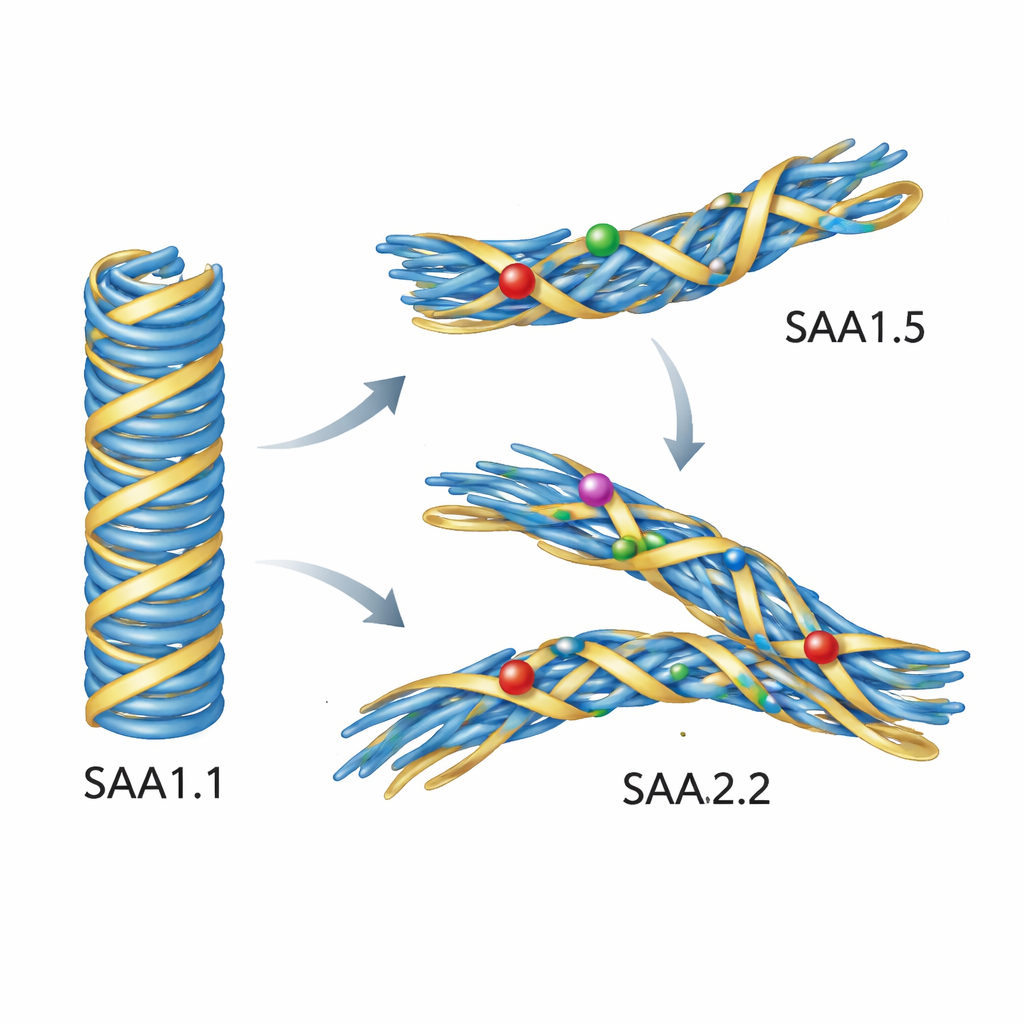
Simulaciones revelan por qué las proteínas mutantes rehúsan la forma dañina
Los experimentos por sí solos no podían mostrar exactamente qué falla a nivel atómico, por lo que los autores recurrieron a simulaciones computacionales detalladas. Partieron de una estructura de alta resolución de una fibrilla AA patógena de ratón construida a partir de SAA1.1 y sustituyeron por ordenador las secuencias de SAA1.5 y SAA2.2. Cuando simularon estas fibrillas en agua a temperatura corporal, el modelo basado en SAA1.1 se mantuvo notablemente estable. En contraste, las fibrillas construidas a partir de SAA1.5 o SAA2.2 se desplazaron y deformaron. Una región clave en forma de lazo del núcleo se movió hacia afuera y aflojó su contacto con el segmento inicial de la proteína, y varias cadenas laterales giraron hacia nuevas orientaciones. Estos sutiles reajustes perturbaron el empaquetamiento compacto que define el plegamiento asociado a la enfermedad. En otras palabras, las secuencias variantes no tenían problema en formar fibrillas en general, pero no podían acomodarse de forma cómoda al plano estructural de la fibrilla AA patógena.
Cómo el diseño de la naturaleza apunta a futuras terapias
En conjunto, el trabajo muestra que las cepas de ratón “resistentes a amiloides” no están protegidas porque su SAA no pueda agregarse en absoluto. Más bien, sus versiones de SAA son incompatibles estructuralmente con la única forma fibrilar concreta que causa la amiloidosis sistémica AA. Las proteínas siguen pudiendo agregarse, pero lo hacen en formas alternativas aparentemente benignas. Se conocen mutaciones protectoras similares en otras enfermedades por plegamiento erróneo de proteínas, incluyendo algunos casos de priones y Alzheimer. Esto sugiere un principio más amplio: modificar una proteína propensa a la enfermedad de modo que no pueda adoptar su arquitectura tóxica —mientras sigue permitiendo su función normal— puede ser suficiente para prevenir la dolencia. A largo plazo, terapias inspiradas en estas variantes naturales “resistentes”, o en fragmentos cortos derivados de ellas, podrían ayudar a desviar a las proteínas de pliegues nocivos hacia formas inocuas.
Cita: Moderer, T., Schnell, A.F., Scheurmann, N.J. et al. Assessment of the biochemical basis underlying the resistance against systemic amyloidosis. Sci Rep 16, 1313 (2026). https://doi.org/10.1038/s41598-026-35297-9
Palabras clave: Amiloidosis AA, amiloide sérico A, plegamiento erróneo de proteínas, resistencia a amiloides, modelos de ratón