Clear Sky Science · es
LASSO estocástico para datos genómicos de dimensión extremadamente alta
Encontrando las agujas en los pajarales genómicos
La biología moderna puede medir decenas de miles de genes a la vez, pero los estudios de pacientes a menudo incluyen solo unos pocos cientos de personas. Ocultos en este desequilibrio hay pequeños conjuntos de genes que realmente importan para predecir el riesgo de enfermedad o la supervivencia. Este artículo presenta “LASSO estocástico”, un método estadístico diseñado para descubrir de forma fiable esos genes clave en océanos de datos genómicos ruidosos, incluso cuando hay muchos más genes que pacientes.
Por qué es tan difícil escoger los genes correctos
Los investigadores suelen apoyarse en herramientas como LASSO, que reducen los efectos de genes poco importantes hacia cero mientras mantienen los más informativos. Sin embargo, las versiones clásicas de LASSO tienen dificultades cuando el número de genes eclipsa al número de muestras, como es habitual en la genómica del cáncer. El LASSO estándar solo puede seleccionar como máximo tantos genes como pacientes hay, y tiende a pasar por alto genes que se comportan de forma similar entre sí. Mejoras anteriores que añaden penalizaciones extra pueden manejar parte de esta correlación, pero también pueden difuminar el significado biológico al forzar que genes relacionados actúen como si todos empujaran los resultados en la misma dirección.
Construyendo muestras aleatorias más limpias
Una solución prometedora es ajustar repetidamente LASSO sobre muchos subconjuntos más pequeños de genes seleccionados al azar y luego combinar los resultados. Aun así, estos enfoques de “bootstrap” sufren de tres problemas: los genes correlacionados pueden cancelarse entre sí, muchos genes rara vez o nunca se muestrean, y la pura aleatoriedad hace que la selección final sea inestable. LASSO estocástico aborda estos problemas directamente con un nuevo esquema de muestreo llamado bootstrap basado en correlación. En lugar de elegir genes al azar, favorece deliberadamente genes que están menos correlacionados con los ya seleccionados, produciendo conjuntos más pequeños de genes que son mucho más independientes. También se asegura de que cada gen se use el mismo número de veces a lo largo de las ejecuciones bootstrap, de modo que ningún gen sea injustamente ignorado. 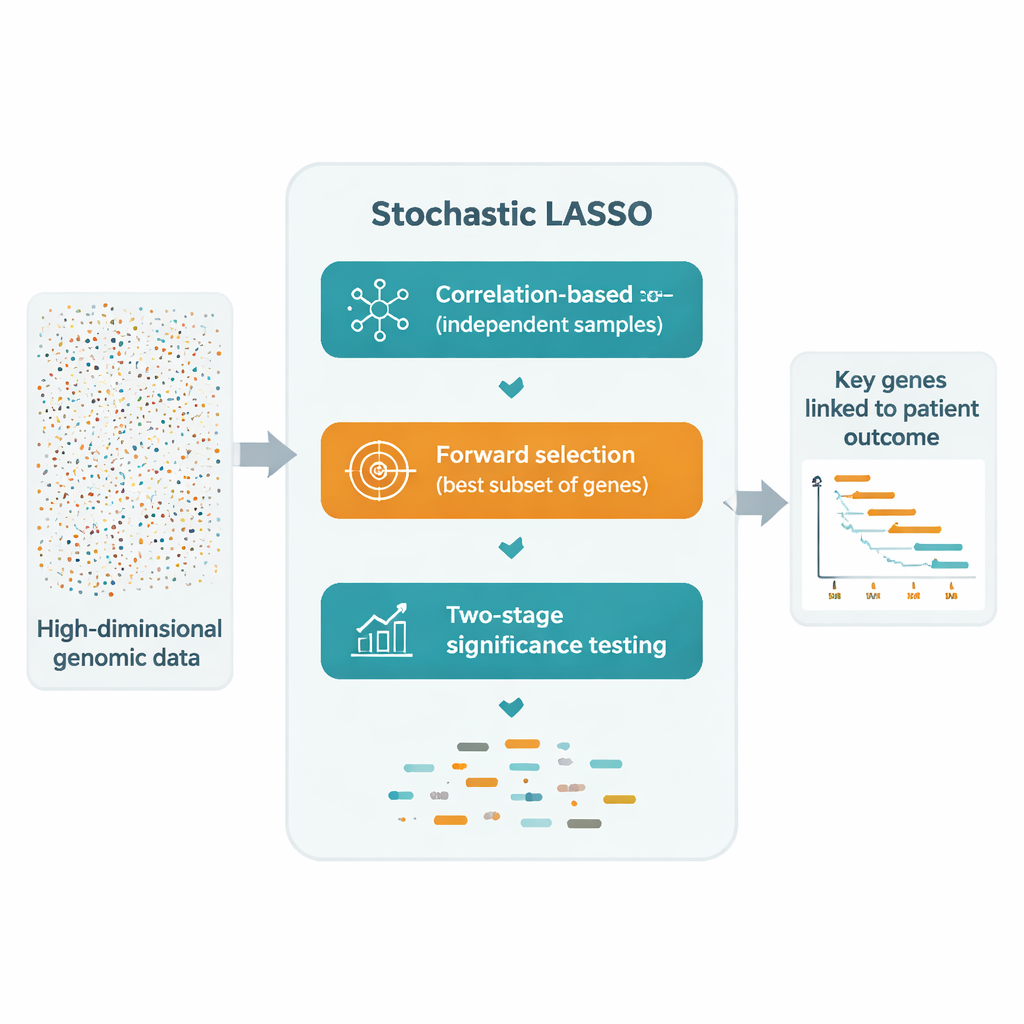
De pistas locales a un conjunto global de genes
Tras construir estos subconjuntos más limpios, LASSO estocástico registra cuán grande es el coeficiente de cada gen a lo largo de todos los ajustes bootstrap. Este efecto absoluto medio se convierte en una “puntuación local” que refleja con qué consistencia aparece importante el gen. En lugar de probar exhaustivamente todas las combinaciones posibles, el método construye modelos candidatos añadiendo genes en orden de sus puntuaciones locales y evalúa qué tan bien cada candidato predice los resultados en datos de validación separados. De este modo, se decide por un conjunto compacto de genes cuyas señales combinadas explican mejor los datos, usando muchas menos pruebas que los métodos secuenciales tradicionales.
Comprobando qué genes realmente importan
Para pasar de “seleccionado con frecuencia” a “estadísticamente convincente”, los autores introducen una prueba t en dos etapas. Primero, verifican si el coeficiente medio de cada gen a través de los bootstrap es claramente distinto de cero, marcándolo como potencialmente significativo. Luego, entre estos candidatos, preguntan si el efecto de cada gen es mayor que el tamaño de efecto típico de todos los candidatos. Solo los genes que superan ambas pruebas se declaran significativos. Debido a que estas pruebas se basan en las muchas estimaciones bootstrap, LASSO estocástico puede identificar con confianza más genes significativos de los que hay pacientes, algo que el LASSO convencional no puede hacer. 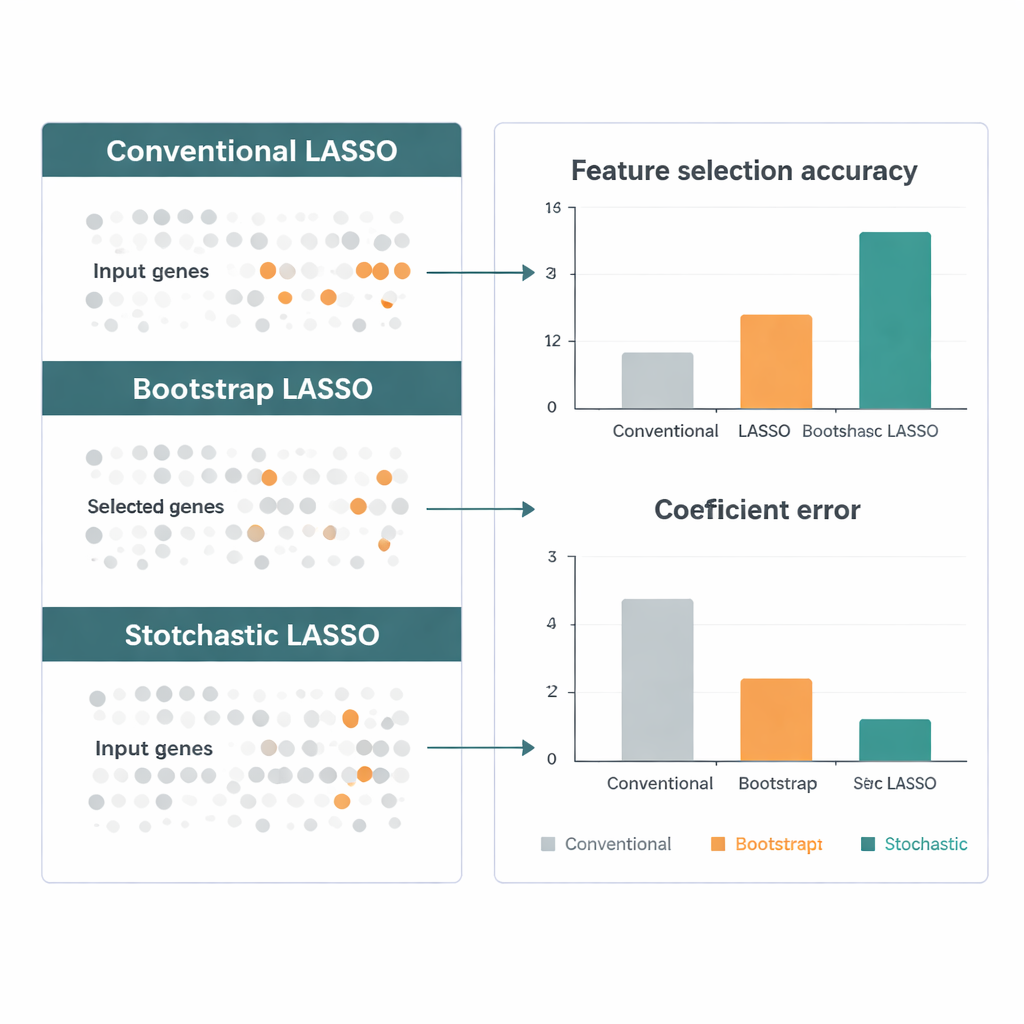
Demostrando su valor en simulaciones y datos de cáncer
Los autores comparan LASSO estocástico con varias variantes líderes de LASSO usando datos simulados diseñados para imitar estudios genómicos reales: muchísimos genes, fuertes correlaciones y señales “verdaderas” conocidas. En múltiples escenarios, el nuevo método encuentra los genes correctos con mayor frecuencia, estima sus efectos con más precisión y se mantiene estable de una ejecución a otra. A continuación aplican el método a datos de expresión génica del The Cancer Genome Atlas para tumores cerebrales, incluidos los agresivos glioblastomas. LASSO estocástico destaca cientos de genes cuya actividad se relaciona con la supervivencia de los pacientes y señala vías biológicas —como rutas de señalización y de metabolismo de fármacos— que tienen respaldo independiente en la literatura, lo que sugiere que el método no solo es estadísticamente más fino sino también biológicamente coherente.
Qué significa esto para pacientes e investigadores
Para no especialistas, el mensaje clave es que LASSO estocástico es un filtro más inteligente para los grandes datos genómicos. Ayuda a los científicos a separar genes genuinamente relacionados con la enfermedad del ruido estadístico, incluso cuando los datos son limitados y los genes están altamente interconectados. Al ofrecer listas de genes y estimaciones de efecto más precisas y estables, puede afinar la búsqueda de biomarcadores, dianas terapéuticas y firmas pronósticas en el cáncer y otras enfermedades complejas. Aunque se demuestra en regresión lineal, el mismo marco puede incorporarse a modelos de supervivencia y problemas de clasificación, ampliando su impacto potencial en la investigación biomédica.
Cita: Baek, B., Jo, J., Kang, M. et al. Stochastic LASSO for extremely high-dimensional genomic data. Sci Rep 16, 5250 (2026). https://doi.org/10.1038/s41598-026-35273-3
Palabras clave: selección de rasgos genómicos, datos de alta dimensionalidad, métodos LASSO, expresión génica en cáncer, descubrimiento de biomarcadores