Clear Sky Science · es
El lactato regula el eje YTHDF2-FTH1 para promover la ferroptosis de los cardiomiocitos y agravar la lesión por isquemia-reperfusión miocárdica
Por qué a los pacientes cardíacos les importa esta química
Cuando los médicos reabren una arteria coronaria bloqueada tras un infarto, el flujo de sangre fresca salva músculo pero también puede causar un daño adicional, conocido como lesión por isquemia–reperfusión. Este estudio revela un culpable sorprendente dentro de las células cardíacas: el lactato, un subproducto metabólico común. Los autores muestran que el lactato puede activar un interruptor molecular que empuja a las células cardíacas hacia un tipo específico de muerte celular impulsada por hierro, empeorando la lesión. Comprender esta vía oculta podría apuntar a fármacos nuevos que protejan mejor el corazón durante el tratamiento de emergencia.
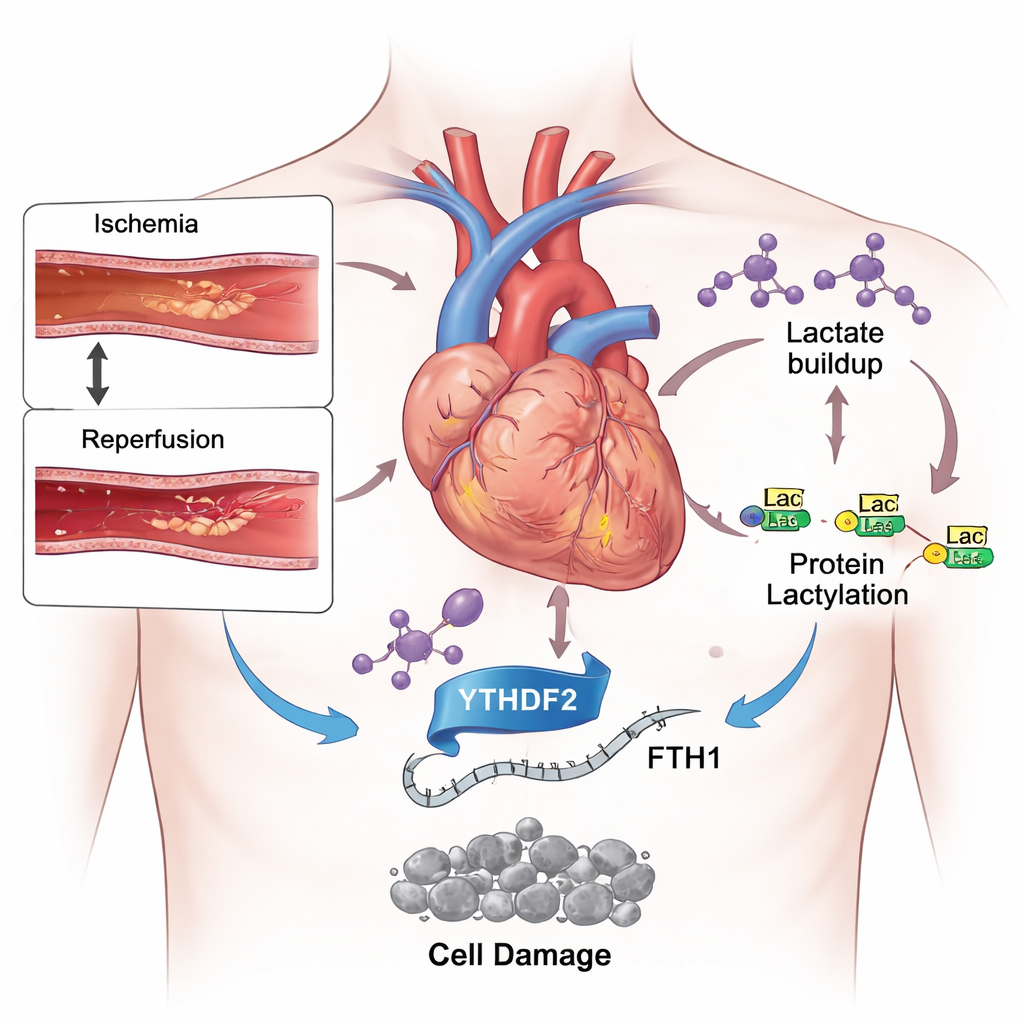
Una espada de doble filo en el tratamiento del infarto
La medicina moderna se ha vuelto muy eficaz para reabrir rápidamente las arterias coronarias obstruidas, limitando el daño inicial de un infarto. Sin embargo, los pacientes aún pueden perder amplias zonas de músculo cardíaco después de restaurarse el flujo sanguíneo. Una razón es que el retorno repentino de oxígeno y nutrientes genera una tormenta de estrés químico dentro de las células cardíacas. Entre varios tipos de muerte celular desencadenados en este contexto, una más reciente llamada ferroptosis ha atraído la atención. A diferencia de formas más familiares como la apoptosis, la ferroptosis depende del hierro y de una oxidación descontrolada de las grasas en las membranas celulares, lo que puede debilitar de forma permanente el corazón.
Cómo el lactato deja de ser solo la "quemazón muscular"
Durante un infarto, el músculo cardíaco privado de oxígeno cambia su uso de combustible hacia la glucólisis, un sistema alternativo que descompone rápidamente el azúcar pero produce grandes cantidades de lactato. Usando ratones sometidos a un breve bloqueo y reablación de una arteria cardíaca, y células cardiacas en cultivo expuestas a hipoxia y posterior reoxigenación, los investigadores hallaron niveles de lactato marcadamente aumentados. Al mismo tiempo, detectaron más de una marca química llamada lactilación en muchas proteínas y en histonas, los andamios que organizan el ADN. Cuando administraron a los animales un fármaco que ralentiza la glucólisis y reduce la producción de lactato, el daño cardíaco disminuyó, los marcadores sanguíneos de lesión cayeron y mejoró el equilibrio entre hierro dañino y antioxidantes protectores. Estos resultados sugieren que el exceso de lactato no es solo un subproducto del estrés, sino un impulsor activo del daño.
Un interruptor molecular que afloja la rienda del hierro
Profundizando, el equipo se centró en YTHDF2, una proteína que lee marcas químicas en el ARN y decide con qué rapidez se destruyen ciertos mensajes. Descubrieron que la isquemia–reperfusión y la adición de lactato aumentaban ambos los niveles de YTHDF2 e incrementaban la lactilación alrededor del gen que lo codifica, amplificando su producción. Uno de los objetivos clave de YTHDF2 resultó ser el ARN de la cadena pesada de la ferritina 1 (FTH1), una pieza central de la jaula celular que almacena hierro. FTH1 normalmente guarda el hierro en una forma segura, evitando que alimente reacciones dañinas. En las células cardíacas estresadas, YTHDF2 se unió con más fuerza al ARN de FTH1 y aceleró su decadencia, dejando a las células con menos jaulas de ferritina, más hierro libre, mayor estrés oxidativo y signos clásicos de ferroptosis.
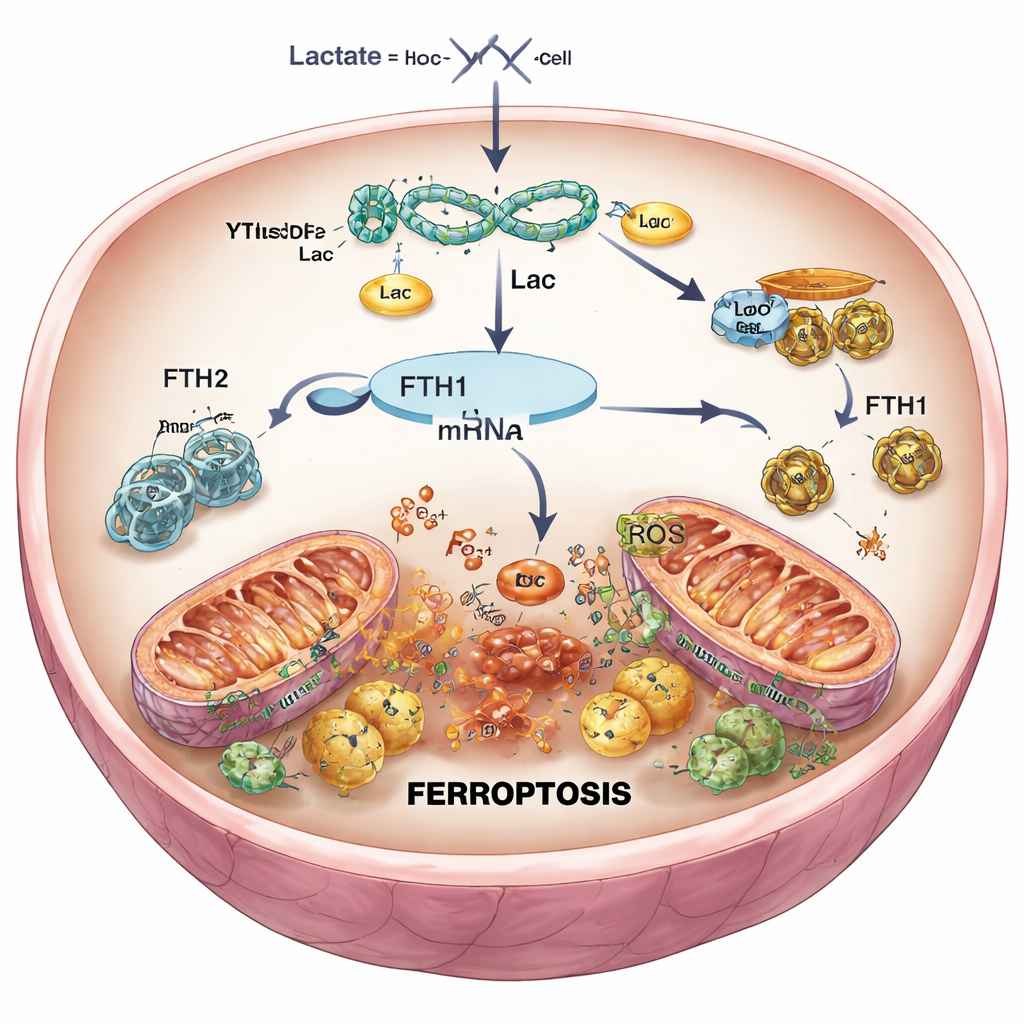
Apagar la señal de muerte en las células cardíacas
Para evaluar causa y efecto, los investigadores usaron herramientas genéticas para reducir selectivamente YTHDF2 en células cardíacas y en ratones. Cuando se silenciaba YTHDF2, los niveles de FTH1 se recuperaban, el hierro y las especies reactivas de oxígeno disminuían, las mitocondrias mantenían una forma más normal y la supervivencia celular global mejoraba tras la reperfusión simulada. En los ratones, menos YTHDF2 se tradujo en cicatrices más pequeñas por el infarto y tejido con mejor aspecto. Sin embargo, cuando FTH1 se redujo simultáneamente, estos beneficios desaparecieron en gran medida: el hierro volvió a aumentar, el daño oxidativo regresó y el tamaño del infarto creció. Esto confirmó que YTHDF2 promueve la ferroptosis principalmente al suprimir FTH1, aflojando el control del hierro dentro de las células cardíacas.
Qué significa esto para futuras terapias cardíacas
Uniendo las piezas, el estudio describe una nueva cadena de acontecimientos: una arteria bloqueada y luego reabierta provoca acumulación de lactato; el lactato aumenta YTHDF2 mediante lactilación; YTHDF2 destruye las instrucciones en ARN para la proteína guardiana del hierro FTH1; y la sobrecarga de hierro resultante desencadena la ferroptosis, profundizando el daño cardíaco. Para los pacientes, el mensaje es esperanzador: esta vía ofrece varios puntos nuevos de intervención. Fármacos que limiten la señalización nociva del lactato, bloqueen la modificación específica de YTHDF2 o preserven la función de FTH1 podrían hacer que la reperfusión de emergencia sea más segura y proteger más músculo cardíaco. Aunque estos hallazgos aún necesitan confirmación en tejidos humanos, abren una vía prometedora hacia tratamientos más suaves y efectivos para los supervivientes de un infarto.
Cita: Xiang, Z., Xiang, B., Ouyang, T. et al. Lactate regulates the YTHDF2-FTH1 axis to promote cardiomyocyte ferroptosis and aggravate myocardial ischemia-reperfusion injury. Sci Rep 16, 4865 (2026). https://doi.org/10.1038/s41598-026-35130-3
Palabras clave: infarto de miocardio, lactato, muerte celular impulsada por hierro, lesión por isquemia-reperfusión, protección de los cardiomiocitos