Clear Sky Science · es
Conjunto de datos virtual mínimo para el ensamblado de novo reproducible de genomas triploides
Por qué importan los genomas con tres copias
Muchas plantas y otros organismos no tienen solo dos copias de cada cromosoma, como los humanos: pueden tener tres o más. Reconstruir esas copias adicionales a partir de datos de secuenciación es sorprendentemente difícil, porque las copias son muy parecidas pero no idénticas. Este artículo presenta un conjunto de datos “virtual” pequeño pero cuidadosamente diseñado que permite a los investigadores probar y comparar programas de ensamblado de genomas ante un problema realista de tres copias (triploide), en condiciones completamente conocidas y reproducibles.
Construir un genoma sustituto simple
En lugar de partir de una planta o animal real, el autor crea primero una secuencia aleatoria de ADN de un millón de bases para servir como plantilla limpia. Esta plantilla se duplica en tres versiones separadas, que representan los tres juegos de cromosomas de un organismo triploide. Para imitar cómo los genomas reales cambian lentamente con el tiempo, el estudio introduce un número fijo de cambios pequeños—sustituciones de una sola base—paso a paso en cada copia. Repetir este proceso a lo largo de 100 pasos produce tríos de genomas que van desde casi idénticos hasta claramente, pero moderadamente, diferentes. Este “gradiente de divergencia” controlado es la columna vertebral del banco de pruebas.
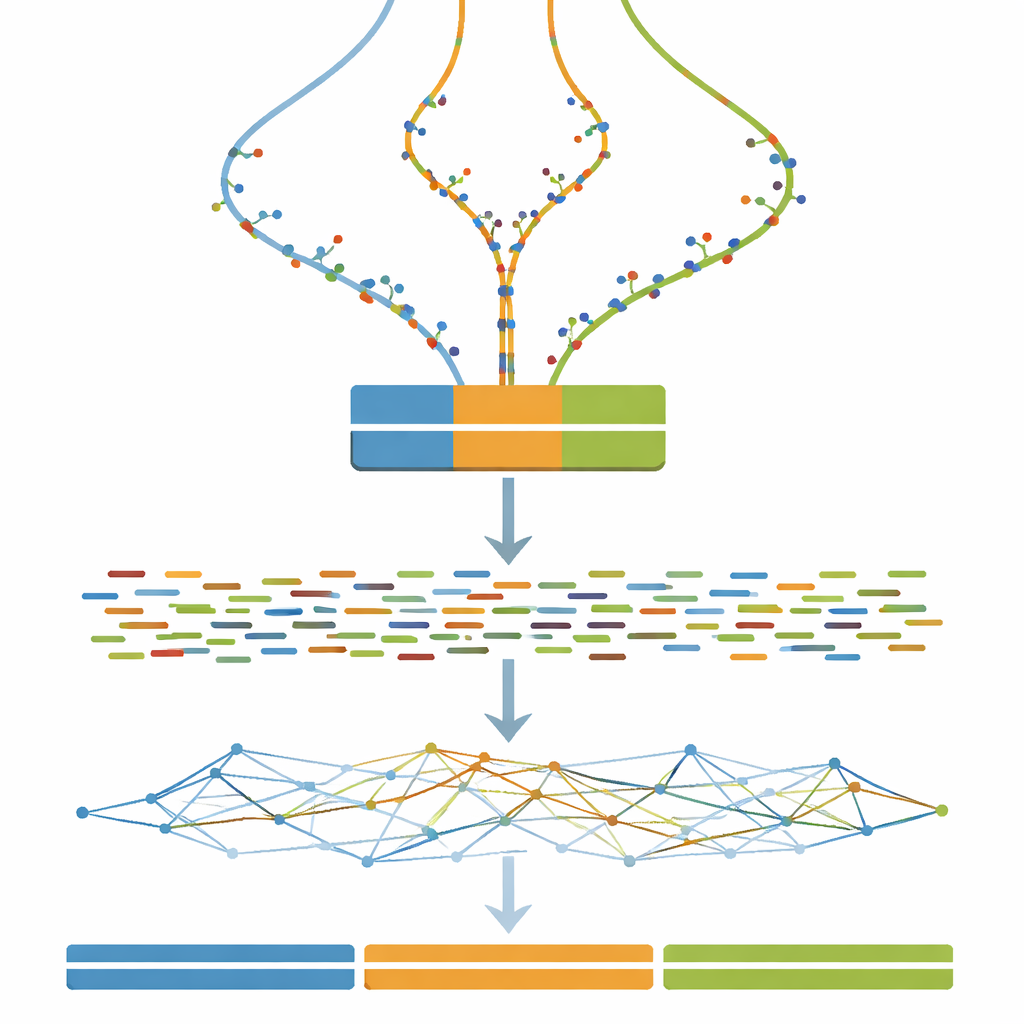
Convertir genomas virtuales en experimentos virtuales
Una vez definido cada genoma triplicado, el siguiente paso es imitar lo que vería una máquina de secuenciación. El estudio utiliza software ampliamente adoptado para simular fragmentos cortos de ADN en pares, similares a los producidos por un secuenciador Illumina, a una profundidad de cobertura constante y bastante alta. Pasos opcionales de limpieza imitan prácticas habituales del mundo real, como corregir errores aleatorios de secuenciación y fusionar pares de lecturas solapadas. Como resultado, cualquiera que use el conjunto de datos puede probar no solo sus algoritmos de ensamblado, sino también cómo las elecciones típicas de preprocesado influyen en los genomas ensamblados finales.
Poner a prueba las estrategias de ensamblado
El núcleo del trabajo es un experimento a gran escala en el que todas las lecturas simuladas se alimentan a un único programa de ensamblado de genomas cambiando solo un ajuste clave: el tamaño del k-mer, un parámetro que controla con qué finura el software «trocea» las lecturas al reconstruir el genoma. Para cada combinación de nivel de divergencia (de 0 a 100 pasos) y tamaño de k-mer (una amplia gama de valores impares), se construye un ensamblado nuevo. Una herramienta de evaluación complementaria mide cuán continuas son las piezas ensambladas, cuántas piezas existen y qué tan cercana es su longitud combinada a la verdad conocida de tres millones de bases. Estas medidas se resumen en mapas de calor que revelan amplias zonas donde los ensamblados colapsan diferentes copias en una sola, se fragmentan en muchas piezas pequeñas o se acercan al ideal de tres contigs largos y precisos.
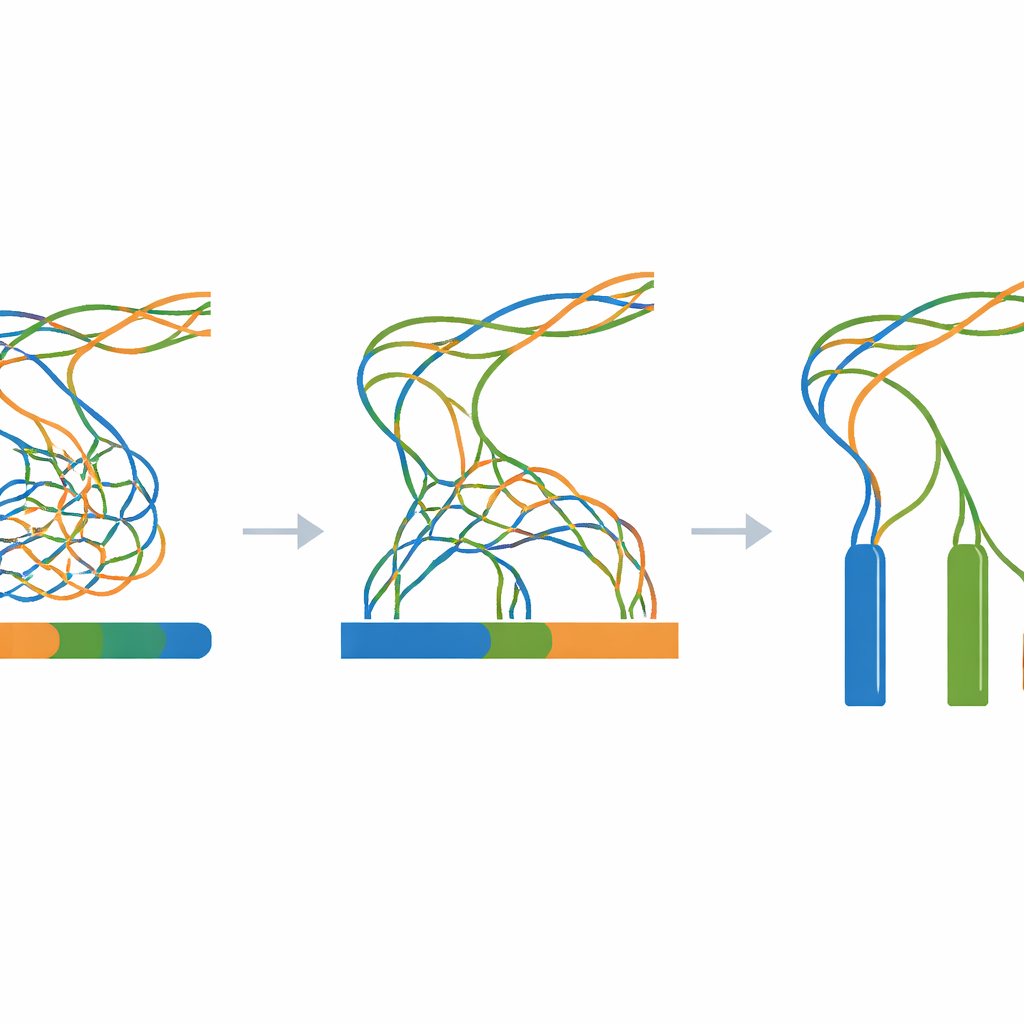
Un referente transparente para genomas complejos
Puesto que cada etapa es sintética y está scriptada—from la plantilla aleatoria inicial hasta los ensamblados finales—los investigadores pueden reproducir todo el flujo de trabajo en cualquier ordenador Linux estándar usando solo herramientas de código abierto. El archivo en Zenodo vinculado en el artículo contiene la plantilla del genoma, todas las secuencias mutadas intermedias, todas las lecturas simuladas y cada resultado de ensamblado, junto con registros y scripts auxiliares sencillos. Comprobaciones técnicas confirman que el proceso de mutación se comporta como se espera, que las lecturas simuladas coinciden con las longitudes y la cobertura solicitadas y que los ensamblados muestran el patrón previsto: fuerte colapso cuando las tres copias son casi idénticas y una separación más clara a medida que se distancian.
Qué significa esto en términos sencillos
En lenguaje cotidiano, este artículo ofrece una pista de prueba controlada para el software que intenta reconstruir tres libros de instrucciones similares a partir de montones de fragmentos barajados. Al aumentar gradualmente cuánto difieren los tres libros y al cambiar sistemáticamente un parámetro clave en el proceso de reconstrucción, el conjunto de datos facilita ver cuándo y cómo los métodos actuales fallan o tienen éxito. Los desarrolladores pueden usarlo para ajustar nuevos algoritmos, mientras que los usuarios pueden comprender mejor qué ajustes funcionan mejor para genomas triploides. Aunque el ADN en sí es artificial, las lecciones que permite—sobre colapso, separación y el impacto de las elecciones de parámetros—son directamente relevantes para los esfuerzos reales de descifrar los genomas complejos de muchas especies importantes.
Cita: Ootsuki, R. Minimum virtual dataset for reproducible triploid de novo genome assembly. Sci Data 13, 382 (2026). https://doi.org/10.1038/s41597-026-06779-2
Palabras clave: ensamblado de genoma triploide, evaluación de poliploidía, conjunto de datos de ADN sintético, ensamblado de novo, optimización de k-mer