Clear Sky Science · es
Conjunto de datos y modelos Open Molecular Crystals 2025 (OMC25)
Por qué importan los grandes volúmenes de datos cristalográficos
Los cristales moleculares están en el centro de muchas tecnologías cotidianas, desde medicamentos y pigmentos hasta electrónica flexible. Su comportamiento depende no solo de los átomos que contienen, sino de cómo innumerables copias de una molécula se empaquetan juntas en un sólido. Predecir ese empaquetamiento y sus consecuencias es notoriamente difícil y lento, y suele exigir costosos cálculos cuántico-mecánicos. Este artículo presenta un nuevo conjunto de datos abierto, llamado Open Molecular Crystals 2025 (OMC25), que reúne decenas de millones de estructuras cristalinas moleculares cuidadosamente simuladas. Está diseñado para ofrecer a los métodos modernos de aprendizaje automático la experiencia rica que necesitan para aprender cómo se comportan estos cristales, con el objetivo a largo plazo de hacer el diseño de cristales más rápido, barato y fiable.

Una biblioteca gigante de cristales modelo
El equipo de OMC25 se propuso construir un extenso “campo de entrenamiento” para modelos informáticos que predicen cómo se ordenan los cristales moleculares y cuán estables son esos arreglos. Reunieron más de 27 millones de estructuras cristalinas construidas a partir de aproximadamente 50 000 moléculas orgánicas diferentes. Cada cristal contiene solo elementos ligeros comunes, como carbono, nitrógeno, oxígeno y algunos halógenos, y puede tener hasta 300 átomos en su unidad repetitiva básica. Para cada estructura registraron no solo las posiciones de los átomos, sino también la energía total del cristal, las fuerzas que actúan sobre cada átomo y el esfuerzo mecánico en la celda unitaria. Estas etiquetas son las que permiten a los modelos de aprendizaje automático conectar patrones en los arreglos atómicos con el comportamiento físico.
De empaquetamientos aleatorios a sólidos realistas
Para poblar esta biblioteca, los autores no se limitaron a copiar cristales experimentales conocidos. En su lugar, usaron una herramienta de código abierto para generar muchas formas diferentes en que una molécula podría empaquetarse en un cristal. Variaron cuántas moléculas hay en la celda unitaria básica y exploraron una amplia gama de simetrías cristalinas. Para cada candidato crearon versiones tanto poco compactas como muy compactas para cubrir situaciones alejadas y cercanas a condiciones realistas. Luego utilizaron un método cuántico de alta calidad, que incluye la atracción sutil entre moléculas, para relajar cada estructura —dejando que los átomos se muevan paso a paso hasta que las fuerzas casi desaparezcan. A lo largo de estos procesos de relajación, muestrearon muchas estructuras intermedias, capturando cómo cambia un cristal mientras se asienta desde una estimación aproximada hasta un arreglo físico probable.
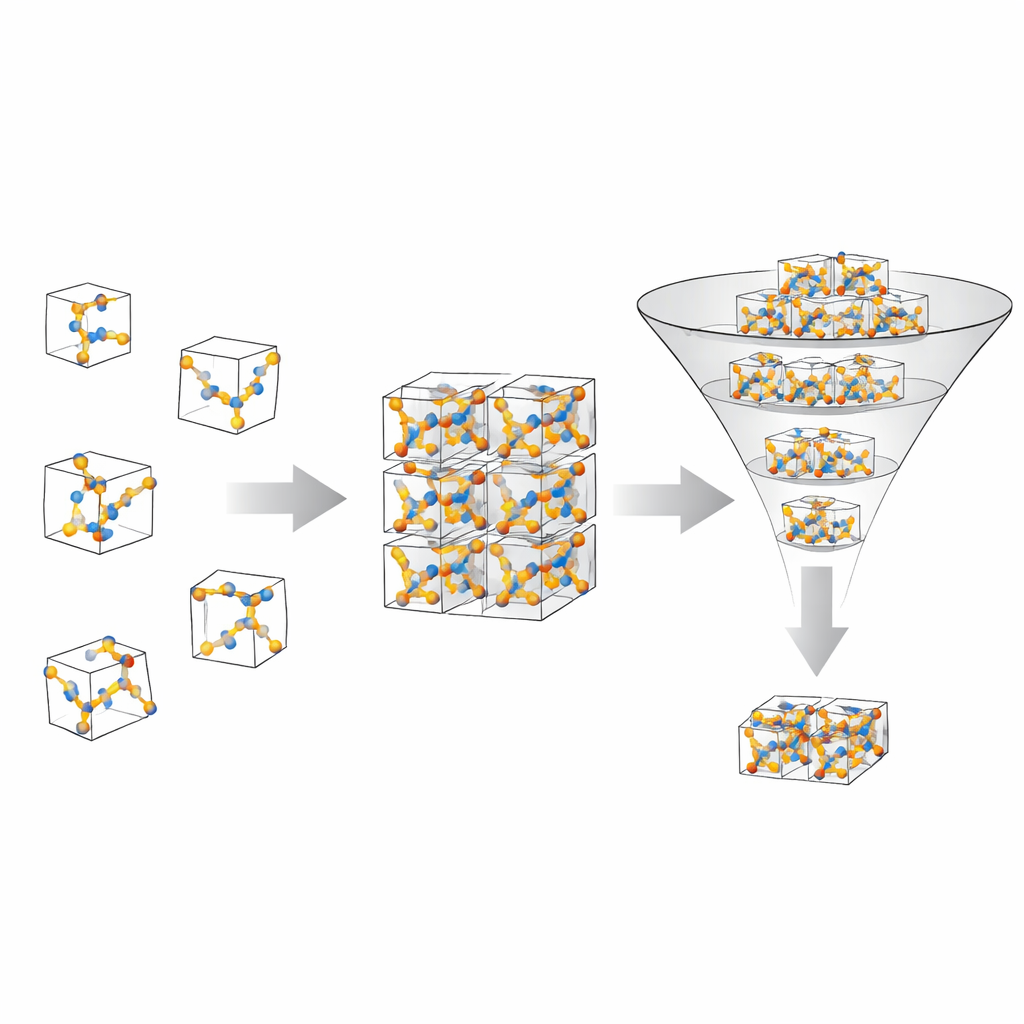
Filtrado cuidadoso y gran variedad
Dado que las estimaciones aleatorias pueden producir situaciones poco realistas, el equipo aplicó filtros estrictos para limpiar los datos. Eliminó cualquier fotograma donde las energías, fuerzas o esfuerzos estuvieran fuera de un rango razonable, o donde las moléculas se rompieran o fusionaran de maneras químicamente poco plausibles. También comprobaron que los volúmenes de celda no saltaran en magnitud hasta el punto de que los parámetros numéricos subyacentes se volvieran poco fiables. El resultado es un conjunto de datos que abarca una enorme variedad de químicas y estilos de empaquetamiento, manteniendo al mínimo los ejemplos no físicos. En comparación con una gran base de datos experimental de cristales, OMC25 contiene una distribución más amplia de simetrías cristalinas y tamaños de celda unitaria, sobresampleando deliberadamente algunos tipos de arreglos para desafiar y enriquecer a los modelos de aprendizaje automático.
Enseñar a las computadoras a predecir cristales
Para probar si OMC25 es realmente útil, los autores entrenaron varios modelos de aprendizaje automático de última generación que operan directamente sobre estructuras atómicas. Estos modelos aprenden a predecir energía, fuerzas y esfuerzo a partir de las posiciones e identidades de los átomos. Cuando se evaluaron con datos reservados de OMC25, alcanzaron errores de predicción muy bajos, lo que demuestra que el conjunto de datos es consistente e informativo. El equipo luego sometió a los modelos a pruebas externas, como reproducir energías y volúmenes cristalinos conocidos y clasificar diferentes formas cristalinas (polimorfos) de la misma molécula. A pesar de haber sido entrenados con datos generados mediante una variante concreta de método cuántico, los modelos compitieron favorablemente en puntos de referencia basados en métodos algo más avanzados, y demostraron ser especialmente fuertes al comparar la estabilidad relativa de distintos empaquetamientos cristalinos.
Qué significa esto para los materiales del futuro
Para los no especialistas, el mensaje clave es que OMC25 ofrece un gran y cuidadosamente curado terreno de práctica donde los modelos de aprendizaje automático pueden “entrenar” con cristales moleculares realistas. En lugar de ejecutar costosos cálculos cuánticos desde cero para cada nueva conjetura cristalina, los investigadores pueden apoyarse cada vez más en modelos rápidos aprendidos a partir de OMC25 para filtrar y refinar estructuras. Esto podría acelerar la búsqueda de mejores formas farmacéuticas, electrónica orgánica más eficiente y materiales especializados mejorados. Aunque el conjunto de datos se centra en una familia particular de cristales y utiliza un nivel de teoría cuántica concreto, establece una base poderosa. Al poner tanto los datos como los modelos de ejemplo a disposición abierta, los autores buscan catalizar esfuerzos más amplios para predecir y diseñar cristales moleculares con la facilidad y rapidez que el aprendizaje automático moderno puede ofrecer.
Cita: Gharakhanyan, V., Barroso-Luque, L., Yang, Y. et al. Open Molecular Crystals 2025 (OMC25) dataset and models. Sci Data 13, 354 (2026). https://doi.org/10.1038/s41597-026-06628-2
Palabras clave: cristales moleculares, potenciales de aprendizaje automático, base de datos de materiales, predicción de estructuras cristalinas, química cuántica