Clear Sky Science · es
AF2BIND: predicción de sitios de unión a pequeñas moléculas usando la representación por pares de AlphaFold2
Encontrar dianas farmacológicas en un mar de proteínas
Los medicamentos modernos a menudo actúan fijándose en pequeños recovecos de la superficie de las proteínas dentro de nuestras células. Sin embargo, incluso con los enormes catálogos de estructuras proteicas disponibles hoy, sigue siendo sorprendentemente difícil predecir de antemano dónde podría adherirse una pequeña molécula —un posible fármaco—. Este estudio presenta AF2BIND, una herramienta computacional simple pero potente que explora el funcionamiento interno de AlphaFold2, el predictor de estructuras proteicas de referencia, para poner de relieve los probables sitios de unión de fármacos en miles de proteínas humanas. Su objetivo es acotar la búsqueda de nuevos medicamentos y revelar puntos funcionales ocultos que los métodos tradicionales pasan por alto.
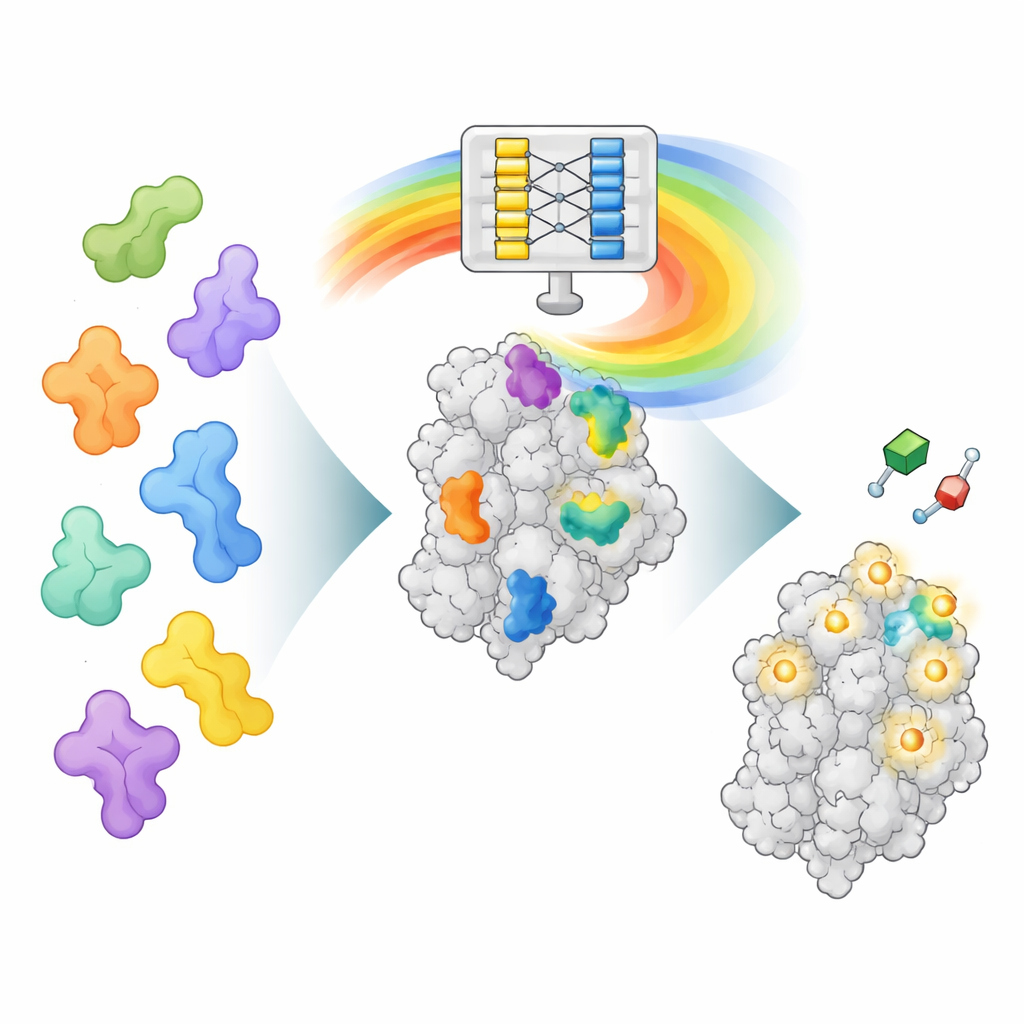
Una nueva forma de leer la “mente” de AlphaFold
AlphaFold2 fue entrenado para predecir cómo una cadena de aminoácidos se pliega en una proteína tridimensional, no para encontrar dónde se unen los fármacos. No obstante, al aprender a plegar proteínas también adquirió patrones ricos sobre cómo interactúan entre sí distintas partes de una proteína. AF2BIND aprovecha una de estas capas internas de datos, llamada representación por pares, que codifica cómo se relaciona en el espacio cada par de posiciones de aminoácidos. Los autores suministran a AlphaFold2 la secuencia de una proteína junto con su estructura de la columna vertebral y además añaden 20 aminoácidos extra, uno de cada tipo, como cadenas “anzuelo” separadas. AlphaFold2 calcula entonces cómo la proteína interactúa con cada residuo anzuelo. Estos patrones de interacción se convierten en la entrada de un modelo de regresión logística muy sencillo que estima, para cada posición en la proteína, la probabilidad de que pertenezca a un sitio de unión a pequeñas moléculas.
Convertir señales ocultas en predicciones prácticas
Entrenar AF2BIND requirió un conjunto cuidadosamente curado de alrededor de 1.900 estructuras proteína–ligando donde las pequeñas moléculas estaban unidas con evidencia experimental de alta calidad. Los investigadores tomaron grandes precauciones para evitar el “engaño” por similitud: dividieron sus datos de modo que las proteínas de prueba no compartieran plegamiento global, secuencia ni siquiera la forma del bolsillo de unión con las empleadas en el entrenamiento. En este riguroso banco de pruebas, la representación por pares de AF2 superó a varias incrustaciones alternativas de redes neuronales, incluidas aquellas basadas únicamente en la secuencia o en diseño de secuencia condicionado por la estructura. Usando solo las características por pares, AF2BIND recuperó aproximadamente dos tercios de los residuos de unión conocidos entre las predicciones mejor clasificadas y mostró un desempeño sólido en métricas de clasificación estándar, manteniéndose además robusto frente a cambios modestos en la forma de la proteína y en las orientaciones de las cadenas laterales.
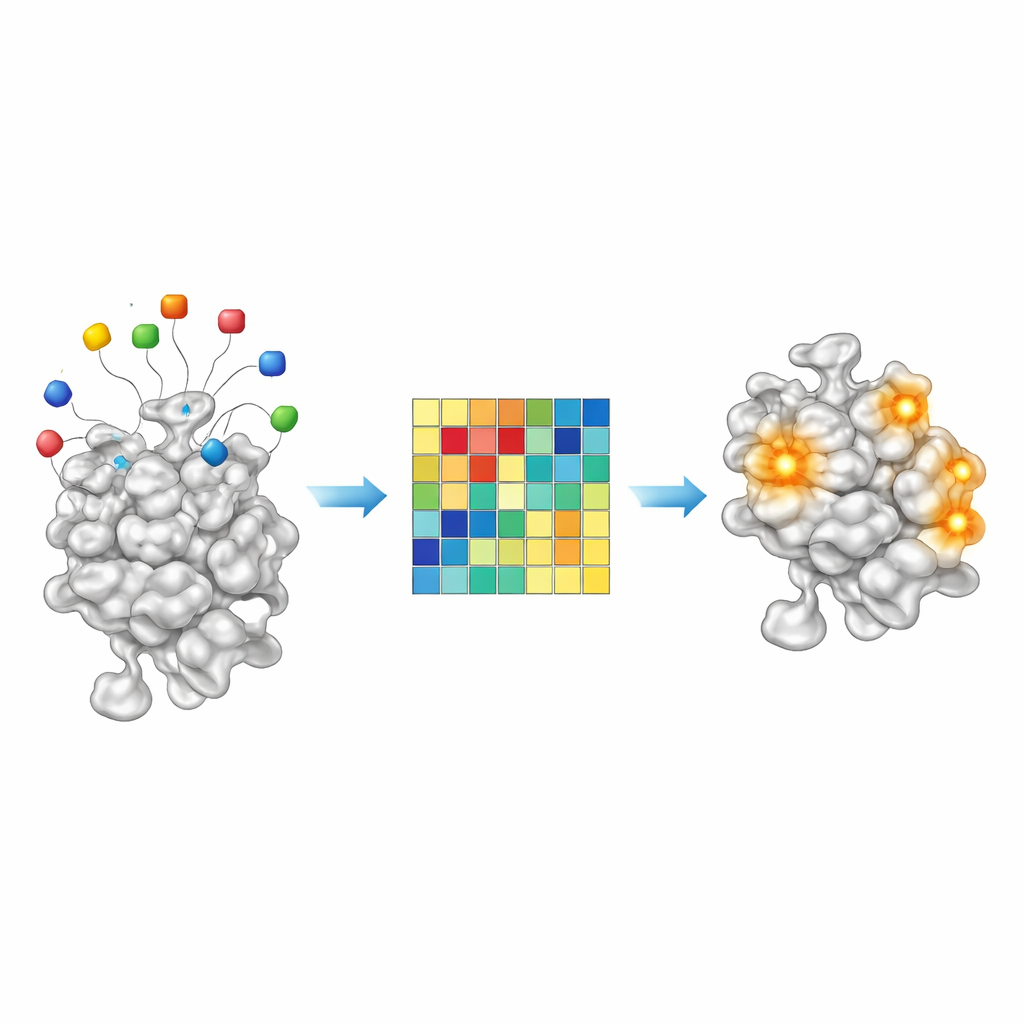
Leer pistas químicas a partir de residuos anzuelo
Puesto que AF2BIND es un modelo lineal sencillo, sus decisiones son inusualmente transparentes para un sistema de IA moderno. Cada uno de los 20 aminoácidos anzuelo aporta una contribución mensurable a la puntuación final de unión en una posición dada de la proteína. Al examinar estas contribuciones en unos 2.000 complejos proteína–ligando, los autores encontraron que ciertas combinaciones de anzuelos se activan con mayor intensidad para ligandos apolares, ricos en carbono, mientras que otras se iluminan para moléculas más polares, atraídas por el agua. En otras palabras, el patrón de activación de los anzuelos actúa como una huella química rudimentaria sobre qué tipos de pequeñas moléculas prefiere un bolsillo dado. Esto sugiere que, en el futuro, enfoques del tipo AF2BIND podrían no solo señalar dónde podría unirse un fármaco, sino también dar pistas sobre la química que encajaría mejor.
Explorar el proteoma humano en busca de nuevos bolsillos
Con su modelo entrenado, el equipo aplicó AF2BIND a las estructuras predichas por AlphaFold de todo el proteoma humano. Tras eliminar regiones de baja confianza y dividir proteínas muy grandes en fragmentos estructurales manejables, agruparon residuos cercanos con puntuaciones altas en candidatos a sitios de unión. AF2BIND predijo más de 20.000 de tales sitios en más de 13.000 proteínas. De manera notable, la mayoría de estos no coincidía con bolsillos inferidos por métodos basados en homología como AlphaFill, que copia ligandos de estructuras cristalográficas relacionadas, ni con un buscador de bolsillos ampliamente usado llamado P2Rank. Muchos de los sitios exclusivos de AF2BIND son más superficiales o difusos que los clásicos bolsillos enterrados y con frecuencia coinciden con regiones que unen péptidos, ARN, ADN u otras proteínas —interfaces que, no obstante, podrían ser atacables por pequeñas moléculas.
Implicaciones para el descubrimiento de fármacos y la enfermedad
Para evaluar cuán prometedores podrían ser estos sitios recién sugeridos para el diseño de fármacos, los autores usaron una herramienta independiente que puntúa la “farmacabilidad” en función del tamaño del bolsillo, su cerramiento y el entorno químico. En promedio, los sitios de AF2BIND obtuvieron puntuaciones por encima de un umbral común para dianas atractivas, incluidos los encontrados en proteínas vinculadas a enfermedades hereditarias. Al cruzar los resultados con experimentos quimioproteómicos que marcan cisteínas reactivas en células, AF2BIND y P2Rank explicaron en conjunto casi la mitad de las regiones observadas como ligandeables, cada método detectando casos que el otro pasó por alto. El trabajo demuestra que las representaciones internas aprendidas por redes de predicción de estructuras pueden reutilizarse para cartografiar sitios probables de unión de fármacos a gran escala, sin conocimiento previo de ningún ligando específico. Para los no especialistas, el mensaje clave es que los mismos avances en IA que predicen formas de proteínas comienzan a revelar dónde y cómo los medicamentos podrían ajustarse mejor a esas formas, acelerando potencialmente la búsqueda de nuevos tratamientos e iluminando puntos de control previamente ocultos en nuestras proteínas.
Cita: Gazizov, A., Lian, A., Goverde, C. et al. AF2BIND: predicting small-molecule binding sites using the pair representation of AlphaFold2. Nat Methods 23, 626–635 (2026). https://doi.org/10.1038/s41592-026-03011-2
Palabras clave: sitios de unión de proteínas, descubrimiento de fármacos, AlphaFold2, biología computacional, bioinformática estructural