Clear Sky Science · es
DECODE: marco común de deconvolución basado en aprendizaje profundo para distintos datos ómicos
Por qué importa esta investigación
La biomedicina moderna está saturada de mediciones de nuestros tejidos: qué genes están activos, qué proteínas están presentes y qué pequeñas moléculas alimentan nuestras células. Sin embargo, la mayoría de estas mediciones se realizan en muestras mezcladas, donde confluyen muchos tipos celulares. El estudio detrás de DECODE presenta un potente marco de inteligencia artificial capaz de desenmarañar esas señales, indicándonos qué células y estados celulares están presentes, incluso entre tipos de datos muy distintos. Esta capacidad podría acelerar la investigación sobre cáncer, inmunidad y enfermedades metabólicas, además de aprovechar mejor las muestras ya almacenadas en biobancos.
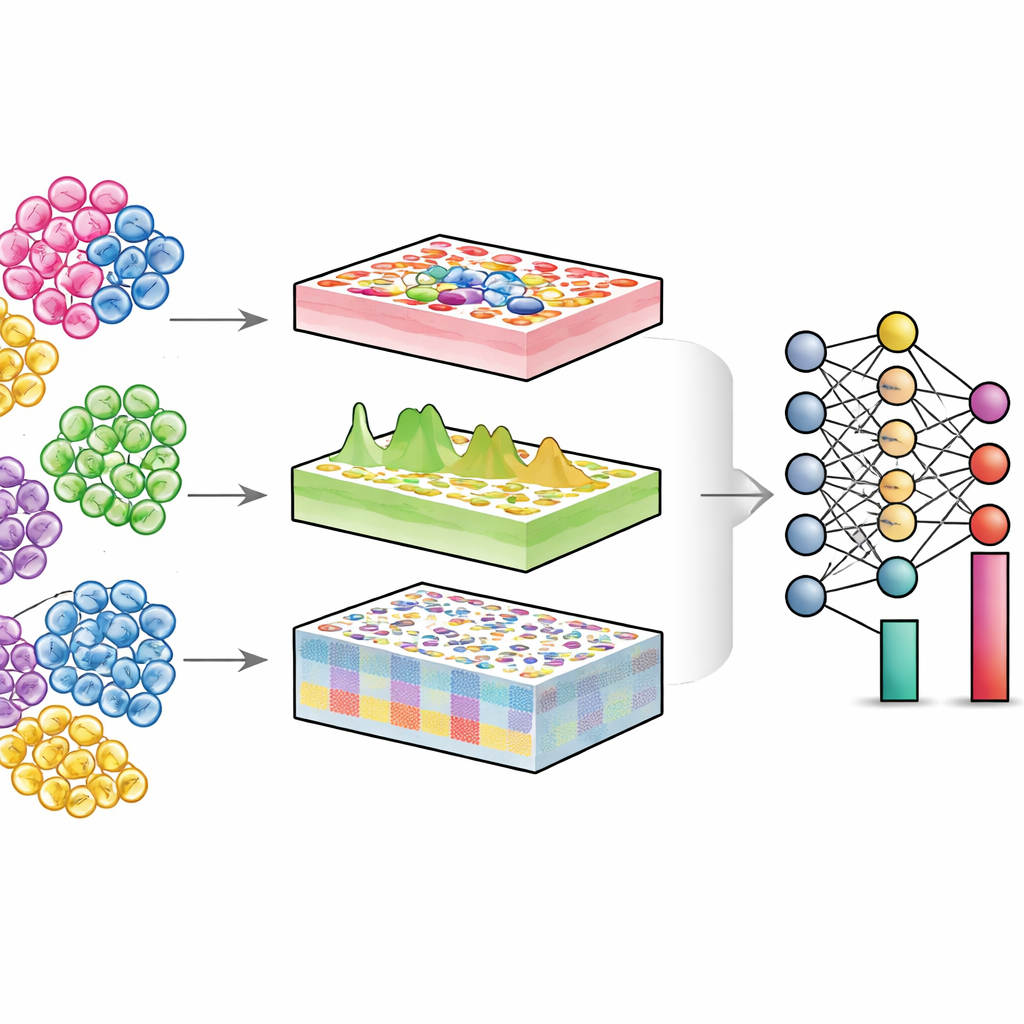
Explorando tejidos mezclados
Cada órgano es una comunidad de distintos tipos celulares: células inmunitarias, células estructurales, células madre y más. En la salud y la enfermedad, lo que cambia a menudo no es solo lo que hace cada célula, sino cuántas hay de cada tipo y en qué estado se encuentran. Las tecnologías de célula única pueden medir células individuales directamente, pero son costosas y técnicamente exigentes, especialmente para grandes cohortes de pacientes o muestras antiguas almacenadas. En cambio, los experimentos convencionales “a granel” mezclan miles o millones de células y proporcionan una señal promedio. Los algoritmos de deconvolución intentan revertir esa mezcla: dados datos a granel y un mapa de referencia de células individuales, estiman la proporción de cada tipo celular en el tejido.
Los límites de las herramientas especializadas
Las herramientas de deconvolución existentes están en su mayoría diseñadas para un único tipo de medición, como la actividad génica (transcriptómica) o las proteínas (proteómica). A menudo asumen comportamientos estadísticos específicos que no se cumplen para otros tipos de datos, y tienen dificultades cuando el tejido a granel contiene tipos celulares ausentes en los datos de referencia. Fuertes efectos de lote —diferencias entre donantes, instrumentos o estados de salud— pueden además difuminar las señales biológicas. Notablemente, no existía un método práctico para metabolómica, el estudio de pequeñas moléculas que con frecuencia están más cerca de los síntomas clínicos. Como resultado, los científicos que analizaban cohortes multiómicas debían compaginar varias herramientas especializadas, cada una con sus particularidades, lo que dificultaba comparar resultados entre estudios y tipos de datos.
Un motor universal de desenredo
DECODE aborda estos retos tratando la deconvolución como un problema flexible de aprendizaje profundo que puede manejar genes, proteínas y metabolitos de forma unificada. Primero, sintetiza “seudotejidos” mezclando digitalmente perfiles de célula única en proporciones aleatorias, creando un conjunto de entrenamiento rico donde se conoce la composición celular real. Una fase de aprendizaje adversario enseña a un codificador a mapear tanto tejidos reales como seudotejidos a una representación compartida donde las diferencias técnicas se minimizan pero se preservan patrones biológicamente relevantes. A continuación, un módulo especial de eliminación de ruido, guiado por aprendizaje contrastivo, aprende a separar las señales tisulares verdaderas del ruido artificial. Este paso hace que DECODE sea robusto frente a tipos celulares faltantes en la referencia y a errores de medición. Finalmente, las características limpiadas se pasan a un módulo de deconvolución que estima abundancias absolutas o relativas de tipos y estados celulares, según cuán completa sea la referencia.
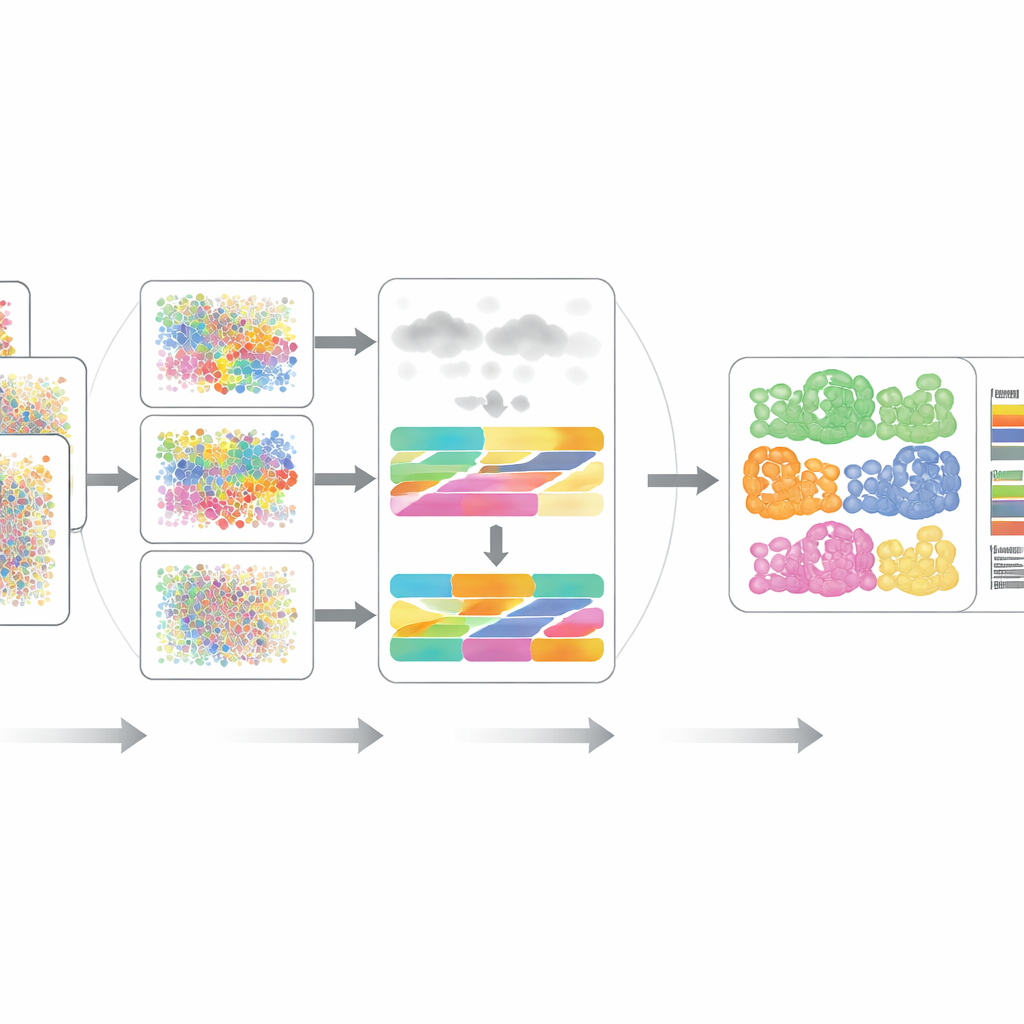
Poniendo DECODE a prueba
Los autores evaluaron rigurosamente DECODE en 15 conjuntos de datos que abarcan siete escenarios realistas, incluidos diferentes donantes, estados de enfermedad, condiciones de salud, plataformas experimentales e incluso mediciones con resolución espacial. En transcriptómica y proteómica, DECODE generalmente igualó o superó a las herramientas de vanguardia en precisión, manteniendo tiempos de cómputo y uso de memoria razonables. De manera crucial, DECODE fue el único método que ofreció resultados fiables en datos metabolómicos, donde hay menos características y distintos tipos celulares pueden parecer engañosamente similares. El marco también demostró habilidad para rastrear estados celulares —como la progresión a lo largo de una trayectoria de desarrollo, fases del ciclo celular o respuestas a tratamientos farmacológicos— en lugar de limitarse a tipos celulares estáticos.
Robusto frente a datos ruidosos e incompletos del mundo real
Los tejidos reales con frecuencia contienen tipos celulares no capturados en referencias de célula única de laboratorio, y el ruido experimental puede distorsionar muchas características a la vez. Los investigadores simularon estos problemas añadiendo tipos celulares desconocidos e introduciendo varias formas de ruido y datos faltantes en transcriptómica, proteómica y metabolómica. En la mayoría de los escenarios, DECODE siguió siendo el método más preciso y, en metabolómica, el único que no colapsó. También demostraron que DECODE ofrece respuestas muy consistentes cuando se aplica a mediciones emparejadas de genes y proteínas de las mismas muestras de células sanguíneas, un requisito clave para comparar cambios en tipos celulares entre capas ómicas en grandes cohortes.
Nuevas ideas biológicas a partir de cohortes multiómicas
Con esta herramienta unificada, el equipo revisó conjuntos de datos de enfermedades complejas. En cáncer de mama, combinaron cohortes transcriptómicas y proteómicas para mostrar cómo las células inmunitarias y las células estromales de soporte cambian entre tumores no metastásicos, tumores primarios en proceso de metastatizar y metástasis cerebrales. Patrones como mayor abundancia de células T y de células de tipo perivascular en lesiones no metastásicas, y aumento de células B en enfermedad avanzada, coinciden y amplían estudios biológicos previos. En hígado de ratón, DECODE integró cohortes transcriptómicas, proteómicas y metabolómicas para seguir cómo hepatocitos, células endoteliales y células inmunitarias residentes cambian bajo distintas dietas y modelos de enfermedad hepática, reproduciendo tendencias conocidas como el aumento de la fracción de células de Kupffer en condiciones inflamatorias.
Qué significa esto de cara al futuro
Para un lector general, el mensaje principal es que DECODE actúa como un prisma inteligente para los datos biomédicos: dado un conjunto de mediciones mezcladas de tejidos, puede separar las contribuciones de muchos tipos y estados celulares, y lo hace de forma fiable en varios tipos de lecturas moleculares. Esto permite a los científicos extraer mucha más información de las cohortes multiómicas y los biobancos existentes sin recopilar nuevos datos de célula única para cada proyecto. Aunque el método aún depende de la calidad y amplitud de las referencias de célula única disponibles, y los recursos de metabolómica siguen siendo limitados, DECODE supone un avance significativo hacia la interpretación rutinaria a nivel celular de estudios humanos a gran escala, con beneficios potenciales para comprender mecanismos de enfermedad y orientar la medicina de precisión.
Cita: Zhao, T., Liu, R., Sun, Y. et al. DECODE: deep learning-based common deconvolution framework for various omics data. Nat Methods 23, 596–608 (2026). https://doi.org/10.1038/s41592-026-03007-y
Palabras clave: deconvolución multiómica, referencia de células individuales, aprendizaje profundo en biología, análisis metabolómico, composición de tipos celulares