Clear Sky Science · es
Variación en las tasas y errores recurrentes de secuencia en filogenias a escala pandémica
Por qué esto importa para futuros brotes
Cuando un nuevo virus se propaga por el mundo, los científicos compiten por leer su código genético y reconstruir su árbol genealógico. Esos árboles ayudan a rastrear cómo surgen las variantes, qué tan rápido se propagan y si las medidas de control funcionan. Pero durante la COVID-19, los laboratorios secuenciaron millones de genomas de SARS‑CoV‑2 tan rápido que errores y peculiaridades ocultas en los datos empezaron a distorsionar la imagen. Este artículo presenta nuevos métodos para limpiar e interpretar conjuntos genéticos tan vastos, ofreciendo visiones más nítidas de cómo un virus pandémico realmente evoluciona y se desplaza entre poblaciones.
El desafío de interpretar millones de genomas
La epidemiología genómica convierte los genomas virales en información práctica para decisiones de salud pública. Para SARS‑CoV‑2, se han compartido más de 20 millones de genomas en todo el mundo. Las herramientas evolutivas tradicionales se diseñaron para problemas más modestos, como comparar genes entre especies, no para manejar millones de secuencias virales casi idénticas que llegan en tiempo real. A esta escala, dos problemas resultan especialmente molestos. Primero, algunos sitios en el genoma viral mutan con mucha más frecuencia que otros, lo que puede hacer que virus no relacionados parezcan extrañamente similares. Segundo, errores técnicos recurrentes en la secuenciación y el procesamiento de datos pueden imitar mutaciones reales. Ambos efectos generan “ecos falsos” en el árbol evolutivo, creando incertidumbre sobre qué ramas y agrupaciones son de fiar.
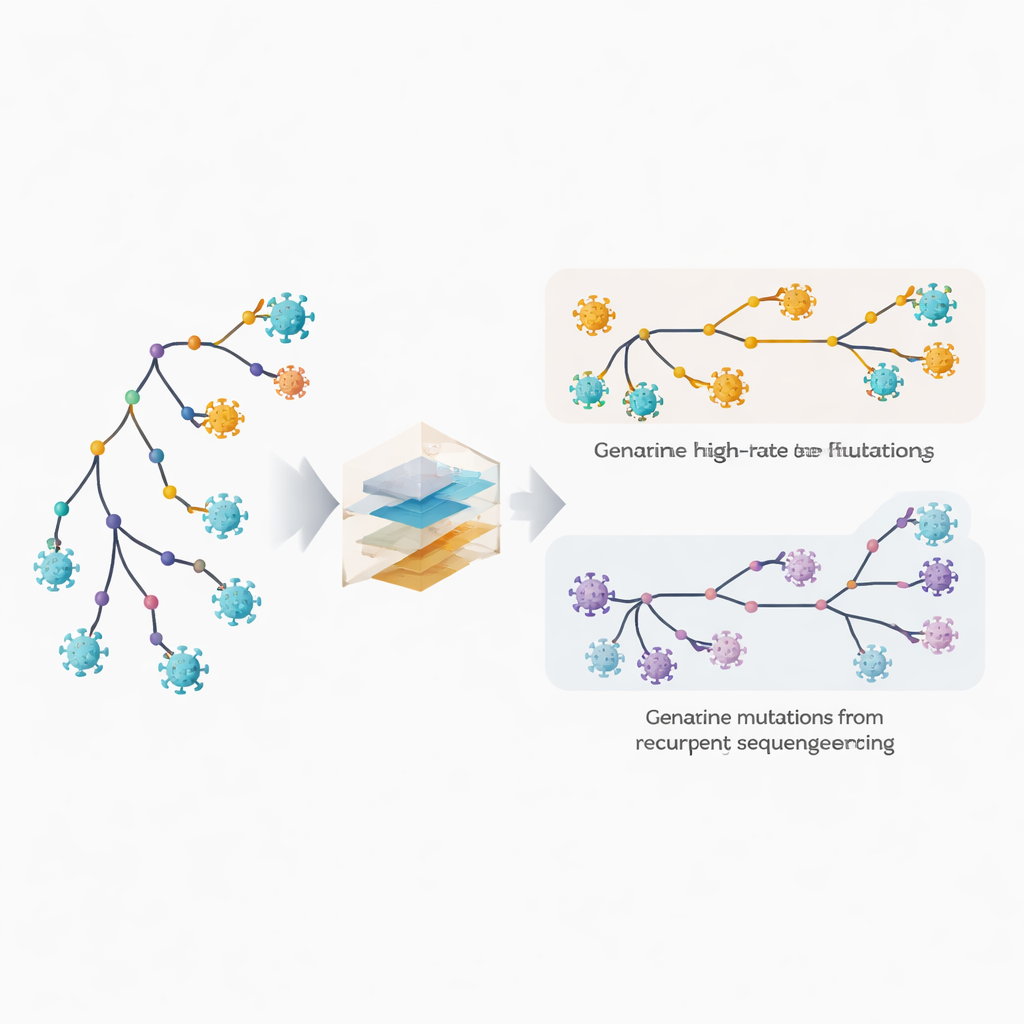
Detectar sitios de cambio rápido y errores ocultos
Los autores amplían su software filogenético, MAPLE, con modelos que tratan cada posición del genoma viral como si tuviera su propio comportamiento. En lugar de asumir un puñado de tasas de mutación medias, el método estima una tasa separada para cada sitio, aprovechando el gran número de genomas disponibles. Al mismo tiempo, permite que cada sitio tenga su propia probabilidad de presentar un error recurrente de secuenciación o de llamada de consenso. La clave es comparar con qué frecuencia aparece un cambio en ramas internas profundas del árbol, que reflejan eventos antiguos compartidos, frente a las puntas más externas, que corresponden a genomas individuales. Las mutaciones biológicas verdaderas tienden a repartirse entre ramas internas y terminales, mientras que los errores técnicos aparecen mayoritariamente en las puntas. Al explotar este patrón, el método puede desenredar la evolución rápida genuina de los errores repetidos.
Algoritmos más rápidos para un árbol de la vida abarrotado
Manejar millones de genomas normalmente requeriría una potencia de cálculo enorme. Para mantener el análisis práctico, el equipo rediseñó cómo MAPLE almacena y actualiza la información de secuencias en el árbol. En lugar de comparar cada genoma con una única referencia fija, el software selecciona “referencias locales” dentro del árbol y registra los genomas cercanos como diferencias relativas a estos anclajes. Esta representación compacta acelera las comparaciones entre partes distantes del árbol. Mejoras adicionales afinan cómo se añaden nuevas muestras a un árbol existente, cómo se ajustan las longitudes de las ramas y cómo se exploran las formas alternativas del árbol, con opciones para ejecutar los pasos más exigentes en paralelo en múltiples núcleos de procesador.
Probar el método y limpiar datos del mundo real
Para comprobar que sus modelos funcionan, los autores primero crearon conjuntos de datos simulados realistas de SARS‑CoV‑2 con patrones de mutación conocidos e errores de secuencia incorporados. En estas pruebas, el nuevo enfoque recuperó árboles evolutivos más fieles y localizó errores individuales con alta precisión, especialmente cuando se incluyeron decenas de miles de genomas o más. Luego se volcaron en datos reales, analizando millones de secuencias de SARS‑CoV‑2 para las que estaban disponibles las lecturas crudas. Al comparar dos tuberías diferentes de construcción de consensos, identificaron posiciones genómicas específicas afectadas repetidamente por artefactos, como problemas de unión de cebadores o llamadas sesgadas por la referencia. Estos sitios sospechosos se enmascararon para análisis posteriores, y se filtraron los genomas que mostraban signos de contaminación o infección mixta, obteniendo un alineamiento curado de más de dos millones de secuencias de alta calidad.
Una imagen global más clara del árbol genealógico del virus
Utilizando el conjunto de datos limpiado, los autores reconstruyeron un árbol filogenético global de SARS‑CoV‑2 y mapearon cómo se relacionan entre sí las principales variantes. Su árbol a veces propone relaciones sutilmente diferentes a las de árboles públicos previos, a menudo de maneras que requieren menos eventos de mutación y encajan mejor con el modelo estadístico. El marco también destaca dónde las etiquetas de linaje pueden ser inconsistentes con la historia genética subyacente, señalando posibles recombinantes o genomas problemáticos para una inspección más detallada. Aunque persisten algunos desafíos —como el sobreajuste cuando los datos son escasos o la influencia de muestras fuertemente contaminadas— el trabajo demuestra que ahora es factible construir árboles evolutivos a escala pandémica más fiables. Para un lector no especializado, la conclusión es que un mejor manejo de errores y de los puntos calientes de mutación conduce a una visión más nítida de cómo los patógenos se propagan y cambian, ayudando a científicos y agencias de salud a responder con mayor rapidez y confianza en futuros brotes.
Cita: De Maio, N., Willemsen, M., Martin, S. et al. Rate variation and recurrent sequence errors in pandemic-scale phylogenetics. Nat Methods 23, 565–573 (2026). https://doi.org/10.1038/s41592-025-02932-8
Palabras clave: genómica de SARS-CoV-2, métodos filogenéticos, errores de secuenciación, variación en la tasa de mutación, epidemiología genómica