Clear Sky Science · es
Modelos de enantioselectividad transferibles a partir de datos escasos
Una forma más inteligente de encontrar el catalizador adecuado
Los químicos a menudo buscan mejores medicamentos y materiales intentando enlazar átomos de carbono en disposiciones tridimensionales muy específicas. Conseguir ese sutil resultado de “mano derecha” frente a “mano izquierda” —conocido como enantioselectividad— suele implicar probar muchos catalizadores metálicos y condiciones de reacción por ensayo y error. Este artículo presenta una manera de usar cantidades relativamente pequeñas de datos experimentales, combinadas con cálculos informáticos rápidos, para predecir qué catalizadores a base de níquel darán la quiralidad deseada en una amplia gama de reacciones, lo que podría ahorrar a los químicos semanas o meses de trabajo en el laboratorio.
Por qué es tan difícil controlar las moléculas quirales
Muchos fármacos y productos naturales existen en formas espejo que pueden comportarse de manera muy diferente en el organismo. Por ello, los catalizadores que favorecen una imagen especular sobre la otra son extremadamente valiosos. Pero diseñar tales catalizadores es complicado. La química cuántica tradicional puede, en principio, calcular qué vía prefiere una reacción; sin embargo, pequeños errores energéticos se traducen en grandes fallos en la predicción de la selectividad, y los cálculos son lentos. Los modelos estadísticos más simples, en cambio, son rápidos pero a menudo ignoran la interacción detallada entre el catalizador metálico y las moléculas reactivas, sobre todo cuando el mecanismo de reacción puede cambiar sutilmente según los diferentes reactivos.
Capturando los momentos clave de una reacción
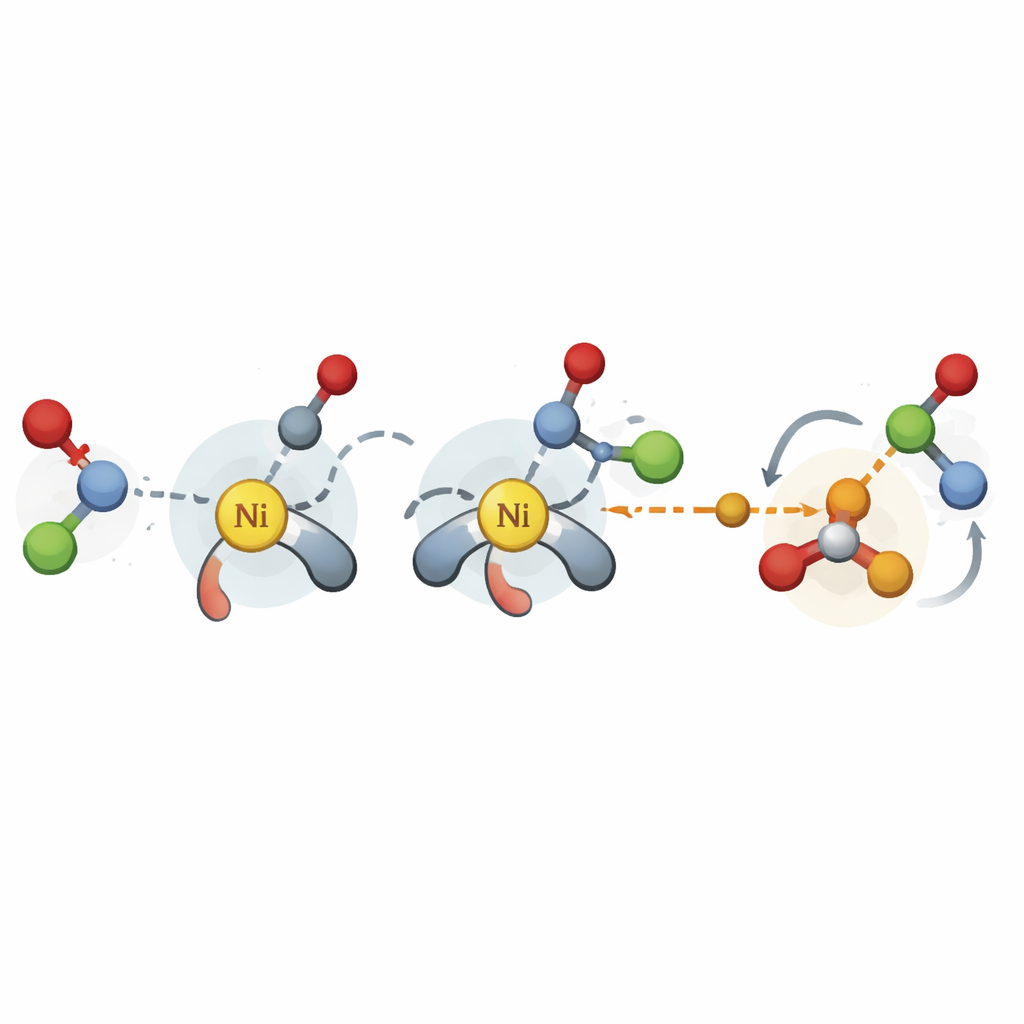
Los autores salvan esta brecha centrándose en las etapas más críticas de una reacción de acoplamiento catalizada por níquel: los pasos en los que se forman nuevos enlaces carbono–carbono y se liberan los productos finales. En lugar de realizar costosas simulaciones de alto nivel, usan un método cuántico simplificado para generar estructuras tridimensionales de los estados de transición e intermedios clave a lo largo de muchas combinaciones posibles de catalizadores y sustratos. A partir de estas estructuras extraen cientos de descriptores físicamente significativos, como cuán congestionado está el entorno del catalizador cerca de ciertos átomos o con qué facilidad pueden moverse los electrones. Estos valores se incorporan luego en modelos de regresión lineal sencillos que conectan características estructurales con la selectividad medida.
Aprender a partir de datos escasos para guiar nuevos experimentos
Un logro central del trabajo es que aprovecha al máximo los datos escasos —las combinaciones limitadas de catalizadores y sustratos que suelen reportarse en un artículo—. En un estudio de caso, el equipo revisita una reacción con níquel que acopla oxiranos de estireno con aril ioduros. Demuestran que los descriptores extraídos del estado de transición más relevante superan a los obtenidos de fragmentos simplificados del catalizador, aunque los cálculos subyacentes son más económicos. Con estos modelos, evalúan virtualmente muchos más ligandos sobre pares de sustratos existentes e identifican nuevas opciones de catalizador que aumentan el exceso enantiomérico para ejemplos particularmente difíciles, todo ello evitando docenas de experimentos innecesarios.
Transferir conocimiento entre distintas reacciones
El enfoque es potente porque puede transferirse entre reacciones de níquel diferentes pero relacionadas. En un segundo conjunto de estudios, los autores combinan datos de varios tipos de reacciones con níquel que todas forman enlaces entre carbonos con hibridación sp3 y socios como grupos arilo o alquenilo, incluso cuando las condiciones exactas o los socios de acoplamiento difieren. Construyendo modelos a partir de los mismos descriptores con significado mecanístico, predicen con éxito la enantioselectividad para nuevos ligandos, nuevas combinaciones de sustratos e incluso una clase totalmente nueva de reacción de formación de enlaces carbono–carbono que no estaba incluida en el conjunto de entrenamiento. El análisis de qué descriptores son más relevantes también sugiere qué paso del ciclo catalítico determina en realidad la quiralidad para cada familia de reacciones.
Ayudar a los químicos a iniciar nuevas reacciones más rápido
En una demostración final, los autores usan su esquema de descriptores junto con una plataforma de optimización bayesiana para diseñar un acoplamiento catalizado por níquel entre acetales benzílicos y aril ioduros que no se había desarrollado de forma asimétrica antes. Partiendo de datos bibliográficos sobre otras reacciones, el modelo recomienda pequeños lotes de ligandos prometedores para probar, concentrándose rápidamente en la clase de mejor rendimiento en solo unas pocas docenas de experimentos. Para un químico, esto significa una herramienta práctica para “arrancar en frío” un nuevo proyecto catalítico: alimentando unos pocos resultados iniciales, el modelo puede sugerir qué ligandos quirales son más propensos a ofrecer alta enantioselectividad. En conjunto, el estudio muestra que características computacionales escogidas con criterio y de bajo coste pueden convertir datos pasados limitados en una guía útil y amplia para construir la próxima generación de reacciones selectivas.
Cita: Gallarati, S., Bucci, E.M., Doyle, A.G. et al. Transferable enantioselectivity models from sparse data. Nature 651, 637–646 (2026). https://doi.org/10.1038/s41586-026-10239-7
Palabras clave: catálisis asimétrica, acoplamiento cruzado con níquel, aprendizaje automático en química, optimización de reacciones, predicción de enantioselectividad