Clear Sky Science · es
Cribados CRISPR bidireccionales descifran un circuito celular fibrótico dependiente de GLIS3
Cuando la curación se convierte en cicatrización perjudicial
Nuestros intestinos están diseñados para repararse tras cada roce e irritación. Pero en enfermedades crónicas como la enfermedad de Crohn y la colitis ulcerosa, este proceso de curación puede descontrolarse, provocando tejido cicatricial grueso y rígido que estrecha el intestino y puede requerir cirugía. Este estudio descubre una conversación oculta entre células inmunitarias y células estructurales del intestino que impulsa esta cicatrización, y señala un interruptor maestro, un gen llamado GLIS3, que podría ofrecer una nueva forma de romper el ciclo.
Una red oculta dentro de intestinos inflamados
Para entender por qué algunos pacientes desarrollan inflamación y fibrosis persistentes, los investigadores crearon un “atlas” celular del intestino humano. Combinaron secuenciación de ARN unicelular, que lee los genes activos en células individuales, con perfilado espacial que sitúa dónde se encuentran esas células en cortes de tejido reales. Usando muestras de personas con enfermedad de Crohn, colitis ulcerosa y controles, cartografiaron más de cuatro millones de células a lo largo de la pared intestinal. Entre esa multitud, destacó un subgrupo de fibroblastos: los fibroblastos asociados a inflamación, o IAF. Estas células se congregaban en áreas de colitis activa y crónica y mostraban una firma génica vinculada a resistencia a las terapias anti‑TNF estándar, lo que sugiere que desempeñan un papel central en la enfermedad de difícil tratamiento. 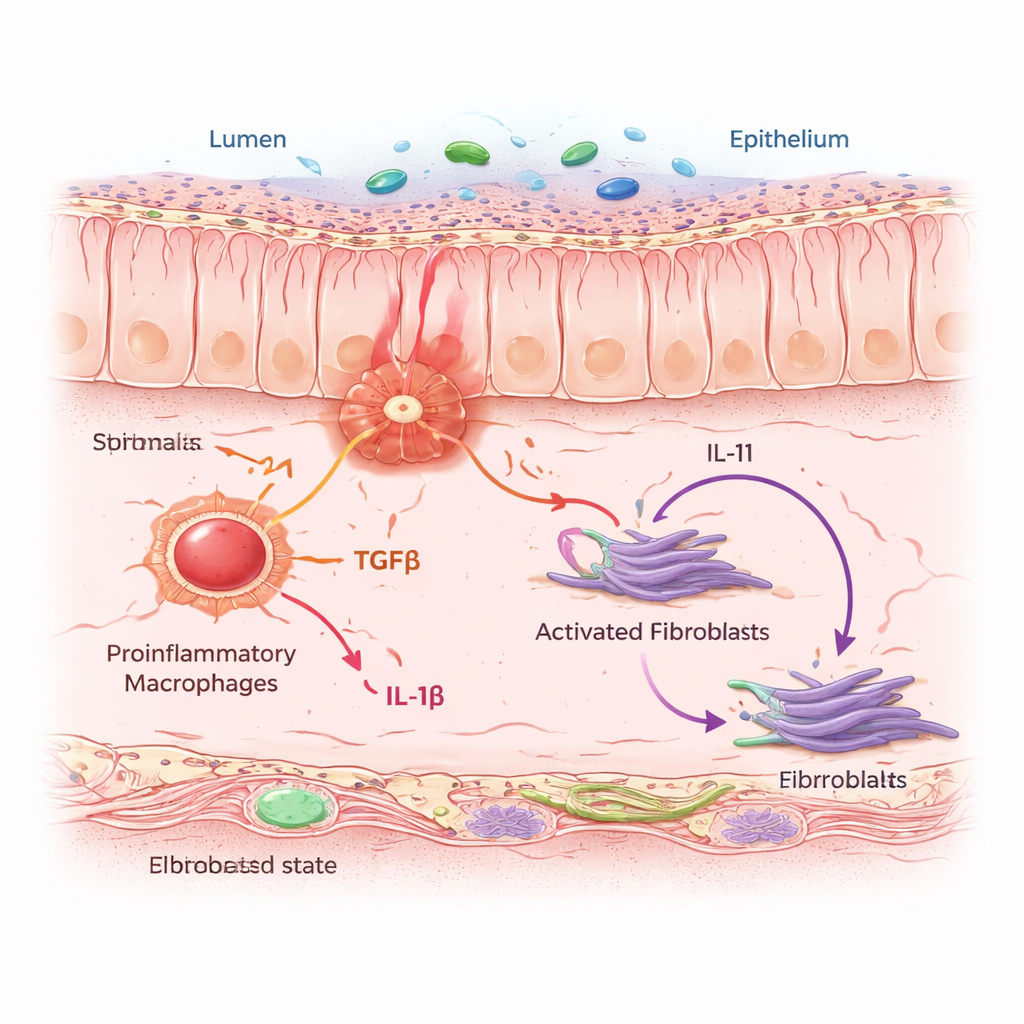
Los macrófagos susurran, los fibroblastos forman cicatrices
Los IAF no actuaban solos. Se agrupaban en “vecindarios” densos en macrófagos proinflamatorios—células inmunitarias que detectan peligro y liberan señales de alarma. Mediante modelos computacionales y experimentos de cocultivo celular, el equipo mostró que cuando los macrófagos se inducen a un estado inflamatorio, secretan dos proteínas mensajeras clave: TGFβ e IL‑1β. Los fibroblastos vecinos escuchan esas señales a través de receptores específicos. Cuando llegan ambas señales simultáneamente, los fibroblastos cambian al estado IAF y comienzan a producir IL‑11, una citocina ya sospechada de promover la fibrosis, además de colágeno y otras proteínas de matriz que engrosan y endurecen la pared intestinal. En ratones sometidos a un régimen de colitis crónica, bloquear IL‑11 o eliminarla selectivamente en fibroblastos redujo la acumulación de colágeno sin impedir la inflamación inicial, demostrando que IL‑11 es un impulsor crucial de la fase de cicatrización.
GLIS3: el interruptor maestro en los fibroblastos fibróticos
Para avanzar de las correlaciones a los mecanismos, los autores emplearon potentes herramientas CRISPR a escala del genoma. Modificaron fibroblastos humanos para que la producción de IL‑11 pudiera supervisarse mediante una etiqueta fluorescente, y luego realizaron cribados paralelos que o bien inactivaban genes o bien los activaban en todo el genoma. Al ordenar las células que producían cantidades inusualmente altas o bajas de IL‑11 tras la estimulación con TGFβ e IL‑1β, identificaron genes que controlan esta respuesta. Entre muchos componentes de señalización, emergió un factor de transcripción como regulador principal: GLIS3. Cuando GLIS3 se desactivó, los fibroblastos produjeron mucho menos IL‑11; cuando se potenció, IL‑11 se disparó. Experimentos adicionales mostraron que GLIS3 se traslada al núcleo del fibroblasto en respuesta a señales de los macrófagos, se une directamente al ADN cerca del gen IL11 y otros, y activa un amplio programa de genes inflamatorios y fibróticos, incluidos colágenos y factores que atraen más células inmunitarias. 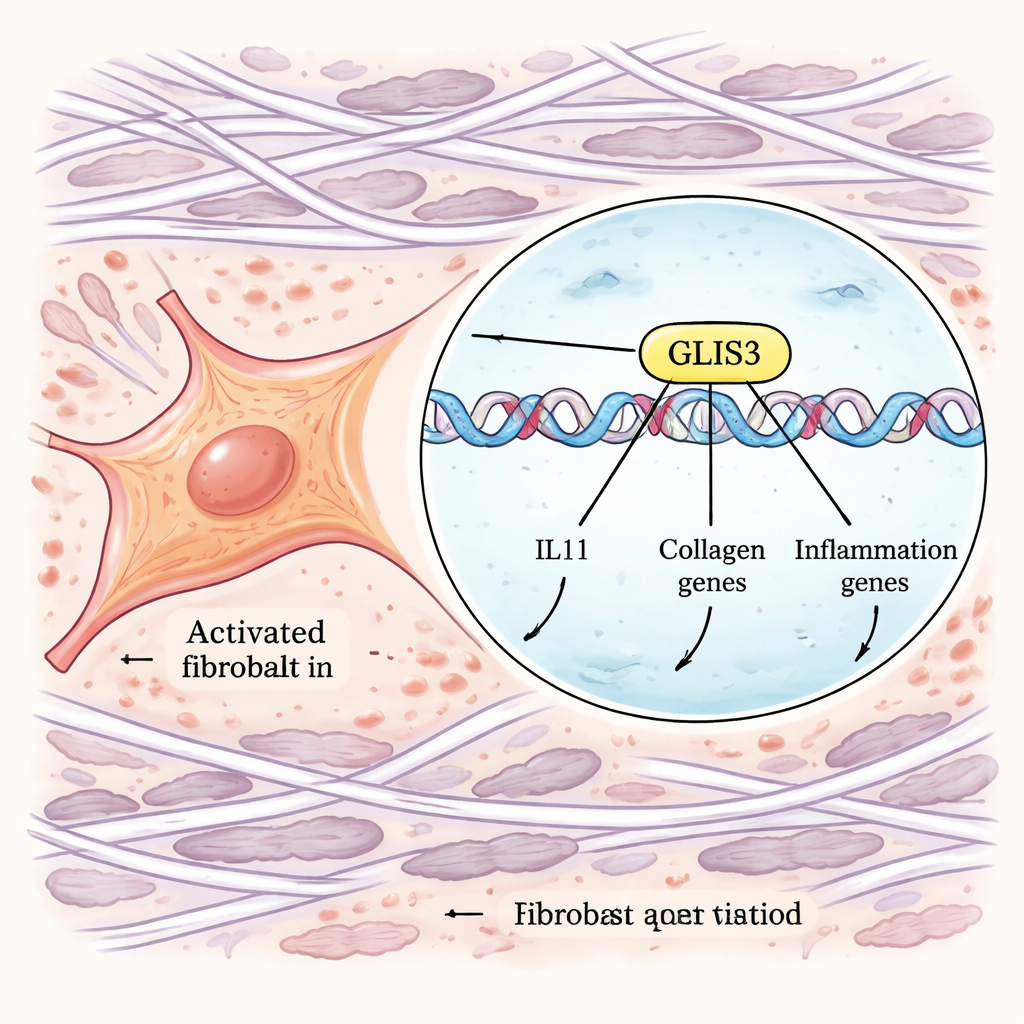
De modelos murinos a la gravedad en pacientes
El equipo preguntó entonces si este programa impulsado por GLIS3 importa en organismos vivos. En ratones, crearon una cepa en la que GLIS3 podía eliminarse solo en fibroblastos. Cuando estos animales fueron sometidos a colitis crónica, desarrollaron menos cicatrización intestinal, presentaron niveles más bajos de colágeno y expresión de genes fibróticos, y mostraron reducción de la inflamación en comparación con ratones normales. El mapeo espacial confirmó que los ratones deficientes en GLIS3 tenían menos fibroblastos productores de IL‑11 y menos macrófagos activados y neutrófilos cercanos, lo que indica que interrumpir GLIS3 debilita todo el circuito inflamatorio‑fibrótico. Al pasar a una gran cohorte pediátrica de colitis ulcerosa, los autores destilaron una “firma” de GLIS3 de 50 genes y encontraron que su actividad en biopsias de colon seguía de cerca la gravedad de la enfermedad y la abundancia de IAFs y macrófagos activados, vinculando esta vía directamente con los resultados en pacientes.
Romper el ciclo de inflamación y cicatrización
Para quienes no son especialistas, la conclusión es que este trabajo revela un bucle autorreforzante: los macrófagos inflamatorios activan a los fibroblastos para que se conviertan en IAFs formadores de cicatrices; estos IAFs, bajo el control de GLIS3, vierten IL‑11, colágeno y otros factores que remodelan el tejido y atraen más células inflamatorias. Los fármacos estándar que suprimen ampliamente el sistema inmune pueden no interrumpir por completo este bucle, lo que ayuda a explicar por qué muchos pacientes acaban fallando a las terapias existentes. Al identificar a GLIS3 y el estado de fibroblasto productor de IL‑11 como nodos centrales en el circuito inflamación‑fibrosis, este estudio señala hacia estrategias más dirigidas—orientadas a los fibroblastos en lugar de solo a las células inmunitarias—que podrían algún día prevenir o revertir la cicatrización en la enfermedad inflamatoria intestinal y posiblemente en otras condiciones inflamatorias crónicas.
Cita: Pokatayev, V., Jaiswal, A., Shih, A.R. et al. Bidirectional CRISPR screens decode a GLIS3-dependent fibrotic cell circuit. Nature 650, 997–1006 (2026). https://doi.org/10.1038/s41586-025-09907-x
Palabras clave: enfermedad inflamatoria intestinal, fibrosis intestinal, fibroblastos, macrófagos, GLIS3