Clear Sky Science · es
Variantes en MTNAP1 subyacen a un trastorno neurodegenerativo al perjudicar la estabilidad mitocondrial
Por qué esta historia importa para la salud cerebral
Muchas familias sufren la angustia de ver a un niño perder lentamente habilidades del desarrollo sin un diagnóstico claro. Este estudio descubre una nueva causa genética de ese tipo de cuadro, trazando el proceso desde un solo gen defectuoso hasta las “centrales eléctricas” dañadas dentro de las células cerebrales y, finalmente, la reducción del tejido cerebral. Comprender esta cadena de acontecimientos no solo aporta respuestas a las familias afectadas, sino que también afina nuestra visión sobre lo frágiles que son realmente los sistemas energéticos del cerebro.
Un trastorno cerebral infantil recién reconocido
Los investigadores estudiaron a tres niños de dos familias no emparentadas que presentaban problemas del desarrollo de inicio temprano. Eran pequeños para su edad, aprendieron a sentarse, caminar y hablar más tarde de lo esperado y luego fueron perdiendo gradualmente algunas de esas capacidades. Todos desarrollaron dificultades motoras como marcha inestable, rigidez muscular o tono muscular bajo, y convulsiones. Las exploraciones cerebrales mostraron un patrón consistente: el tejido tanto del cerebro principal (cerebro) como del “cerebelo” en la parte posterior se adelgazaba con el tiempo, y un puente clave de fibras nerviosas, el cuerpo calloso, era inusualmente delgado. Estas características apuntaban a una pérdida progresiva de neuronas más que a una lesión única al nacimiento.
Un gen diminuto con grandes consecuencias
Para buscar una causa heredada, el equipo secuenció todos los genes codificantes de proteínas en los niños afectados y en sus padres. Se centraron en un gen llamado MTNAP1, que ayuda a organizar el ADN dentro de las mitocondrias, las fábricas de energía de la célula. Cada niño portaba dos copias defectuosas de MTNAP1, una procedente de cada progenitor sano portador. En dos hermanos, un cambio de una “letra” en el gen intercambió un aminoácido por otro, distorsionando sutilmente su forma. En el tercer niño, una señal de parada temprana en el gen probablemente impidió la síntesis de la proteína por completo. Estos cambios no se observaron en grandes bases de datos poblacionales, lo que refuerza la hipótesis de que son variantes raras y dañinas en lugar de rasgos inofensivos.
Centrales eléctricas bajo estrés
Seguidamente, los científicos examinaron células de la piel tomadas de los niños y las compararon con células de individuos sanos. Al microscopio, las células normales mostraban mitocondrias largas y filiformes formando una red conectada, mientras que las células de los niños contenían mitocondrias cortas, fragmentadas y amontonadas. Cuando los investigadores redujeron experimentalmente los niveles de MTNAP1 en una línea celular humana con carácter neuronal, observaron la misma ruptura de la red mitocondrial, confirmando que la pérdida de esta proteína por sí sola puede alterar su estructura. Las mediciones de la actividad mitocondrial revelaron que pasos clave en la producción de energía estaban debilitados, y las células generaban exceso de especies reactivas de oxígeno —subproductos oxidativos dañinos que actúan como óxido molecular. Las células estresadas dejaron de dividirse correctamente, se acumularon en una fase de reposo y activaron marcadores de envejecimiento prematuro.
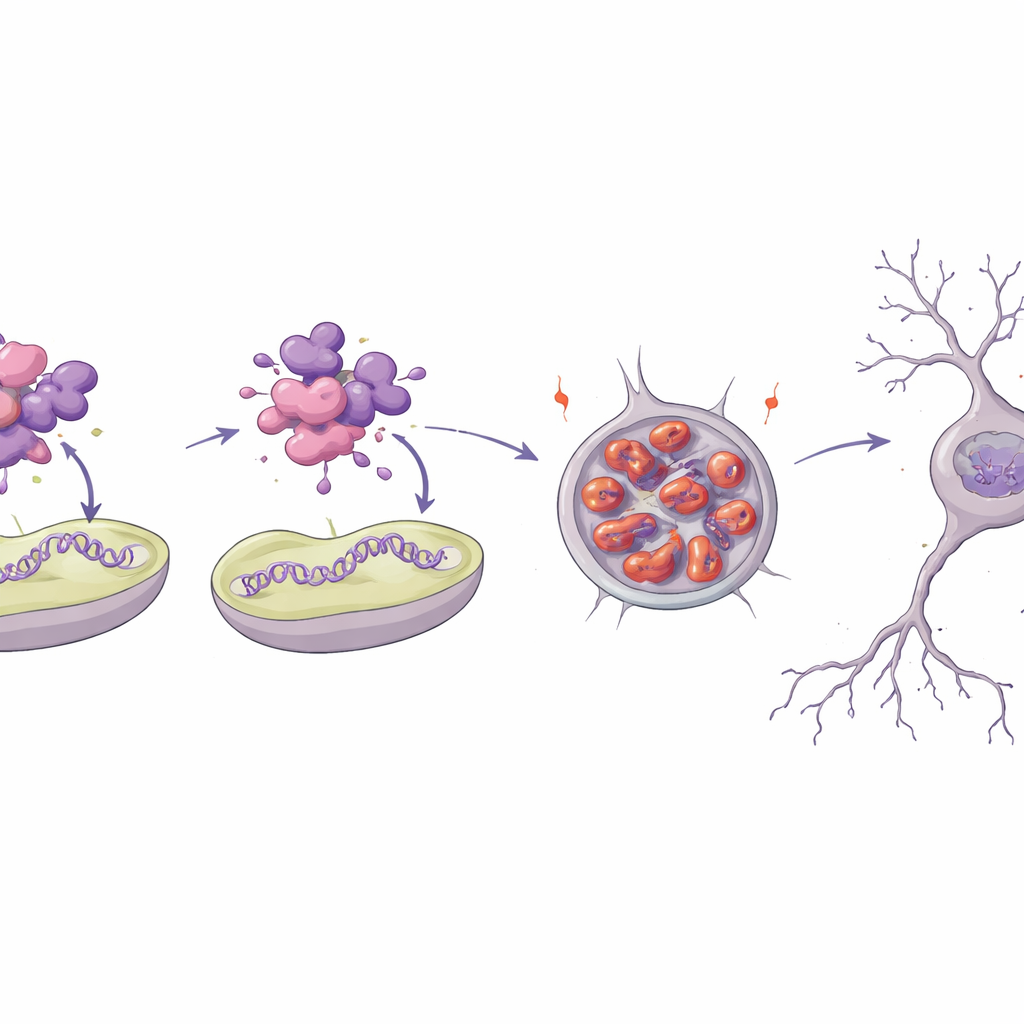
Cómo un único cambio deshilacha una proteína vital
Para entender por qué una de las variantes es tan dañina, el equipo modeló la estructura 3D de la proteína MTNAP1 y la recreó en el laboratorio. El aminoácido sustituido se sitúa en una región helicoidal muy compacta que normalmente ayuda a la proteína a interactuar con el ADN mitocondrial y con la membrana interna. Simulaciones por ordenador y pruebas biofísicas mostraron que la proteína mutante es menos estable, pierde gran parte de su estructura ordenada y tiende a formar agregados. En experimentos de tubo de ensayo, la proteína normal se unía con fuerza a fragmentos cortos de ADN mitocondrial y a superficies de membrana artificiales, mientras que la mutante casi no interactuaba y, en su lugar, se ensamblaba en agregados tipo amiloide. Cuando se introdujo en células con rasgos neuronales, la mutante se acumuló con el tiempo en grandes grumos perinucleares, señal de que los sistemas de control de calidad proteica estaban desbordados.
De las mitocondrias dañadas a un cerebro que falla
Al ensamblar las piezas, el estudio propone un modelo por etapas: MTNAP1 defectuoso debilita el andamiaje que ayuda a organizar el ADN mitocondrial y anclarlo a la membrana interna; esto desestabiliza las mitocondrias, provocando su fragmentación y pérdida de capacidad para generar energía de forma eficiente; el aumento del estrés oxidativo y las señales de envejecimiento prematuro hacen que las neuronas sean especialmente vulnerables, porque tienen demandas energéticas altas y constantes y capacidad limitada para renovarse. En el cerebro en desarrollo, esta crisis energética lenta y continua se traduce en hitos del desarrollo estancados, pérdida de habilidades aprendidas y disminución gradual de regiones cerebrales clave. Aunque se necesitan más pacientes y estudios en animales para mapear completamente el síndrome, este trabajo sitúa con firmeza a MTNAP1 como un guardián crucial de la estabilidad mitocondrial y subraya la organización del ADN mitocondrial como un pilar central del desarrollo cerebral sano.
Cita: Kumar, A., Saha, S., Nasir, N. et al. Variants in MTNAP1 underlie a neurodegenerative disorder by impairing mitochondrial stability. npj Genom. Med. 11, 19 (2026). https://doi.org/10.1038/s41525-026-00554-3
Palabras clave: mitocondrias, neurodegeneración, genética pediátrica, ADN mitocondrial, desplegamiento proteico