Clear Sky Science · es
Mezcla espacial eficiente y precisa de potenciales interatómicos aprendidos por máquina para la ciencia de materiales
Por qué importan las simulaciones atómicas más rápidas
Diseñar mejores materiales para tecnologías como la fusión nuclear, la microelectrónica y las aleaciones estructurales depende cada vez más de simulaciones por ordenador que siguen cómo se mueven e interactúan los átomos. Los métodos más precisos toman ideas de la física cuántica, pero son tan exigentes computacionalmente que solo son prácticos para tamaños de sistema y escalas temporales modestos. Este artículo presenta ML‑MIX, una técnica y paquete de software que permite a los investigadores mantener la precisión cercana a la cuántica exactamente donde es necesaria, mientras usan modelos más simples y baratos en el resto. El resultado es un aumento sustancial de la velocidad —a menudo un factor de 4 a 10— sin perder fiabilidad en las predicciones físicas clave.
Mezclando visiones detalladas y simples de los átomos
El núcleo del trabajo es una idea sencilla: no todos los átomos en una simulación necesitan el mismo nivel de atención. Las regiones donde los enlaces se estiran, rompen o reorganizan —como defectos, superficies o partículas implantadas— se benefician de potenciales interatómicos modernos aprendidos por máquina, que imitan la precisión cuántica. Pero los átomos alejados de estos “puntos calientes” mayormente vibran alrededor de posiciones regulares y pueden tratarse con modelos mucho más simples. ML‑MIX proporciona una forma de combinar un modelo preciso pero caro con otro más ligero y “barato” dentro de la misma caja de simulación. Lo hace definiendo una zona núcleo que usa el modelo caro, un buffer circundante donde las fuerzas se mezclan cuidadosamente entre modelos, y una zona exterior de volumen que utiliza solo la descripción barata. 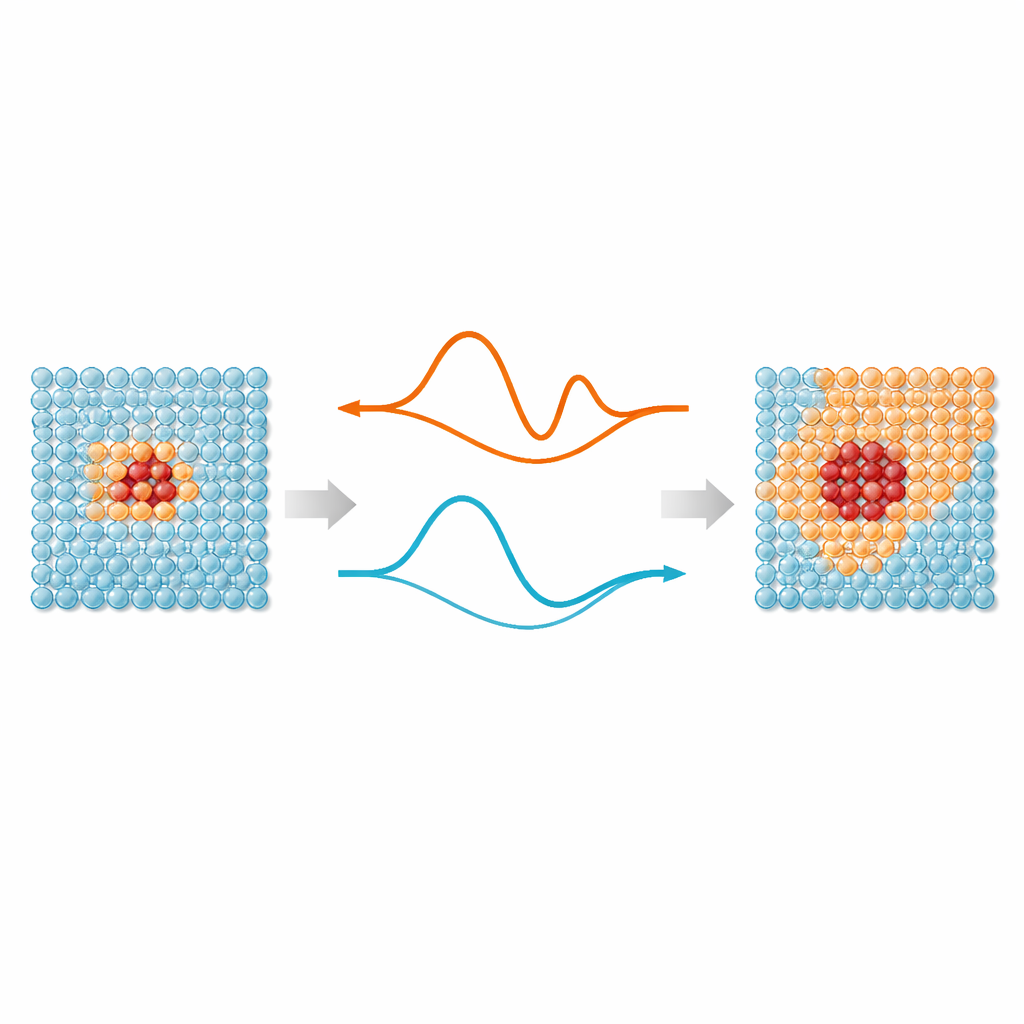
Enseñar a un modelo barato a imitar a uno preciso
Un desafío clave es asegurarse de que el modelo barato se comporte como el preciso donde ambos se encuentran. En lugar de ajustar el modelo barato directamente a un conjunto de datos cuántico enorme y heterogéneo, los autores generan datos “sintéticos” focalizados ejecutando el modelo preciso en las condiciones específicas relevantes para la región de volumen: vibraciones a alta temperatura y cristales ligeramente deformados. Luego ajustan el modelo barato para que coincida con estos datos, imponiendo al mismo tiempo restricciones estrictas sobre propiedades materiales básicas como las constantes elásticas y la separación de la red. Este ajuste con restricciones garantiza que las tensiones y deformaciones a larga distancia coincidan de manera suave a través de la frontera entre los dos modelos, evitando fuerzas artificiales que podrían corromper la dinámica cerca de la interfaz.
Poner el método a prueba
Para comprobar que ML‑MIX funciona realmente, los autores ejecutan una batería de pruebas en sistemas de silicio, hierro y tungsteno. Como ejemplo simple, calculan la barrera de energía para que una vacante —un sitio de la red vacío— en silicio se desplace de una posición a otra. La simulación mixta reproduce el resultado de un cálculo totalmente caro con una discrepancia de una milésima de electrón‑voltio, mientras que se ejecuta aproximadamente cinco veces más rápido. En un escenario más dinámico, estiran un único enlace de silicio en un cristal caliente y miden la fuerza media sobre él. Una simulación que usa solo el modelo barato ya se aproxima sorprendentemente, pero una vez que se añade un pequeño núcleo caro alrededor del enlace estirado, el acuerdo se vuelve estadísticamente indistinguible de la referencia totalmente precisa, con aceleraciones de hasta un factor de aproximadamente 13 en ejecuciones en serie.
Seguir defectos y partículas en movimiento
Pruebas más realistas examinan cómo se mueven los defectos a través de metales. El equipo simula la difusión de un defecto auto‑intersticial en hierro y de átomos de helio dentro del tungsteno. En cada caso, el modelo caro se confina a una pequeña región móvil alrededor del defecto, mientras que el resto del cristal se maneja con el potencial barato. Los coeficientes de difusión resultantes coinciden con los de simulaciones totalmente precisas dentro del error estadístico, incluso cuando una simulación solo con el modelo barato fallaría. Los autores empujan entonces el método a problemas de mayor escala y relevancia científica en tungsteno, un material candidato principal para reactores de fusión. Modelan el movimiento de dislocaciones de tornillo —defectos lineales que controlan la deformación plástica— y la implantación de átomos de helio en una superficie de tungsteno caliente. En ambos casos, ML‑MIX reproduce los resultados de solo‑caro mientras reduce el coste computacional por factores de alrededor de cuatro a once. 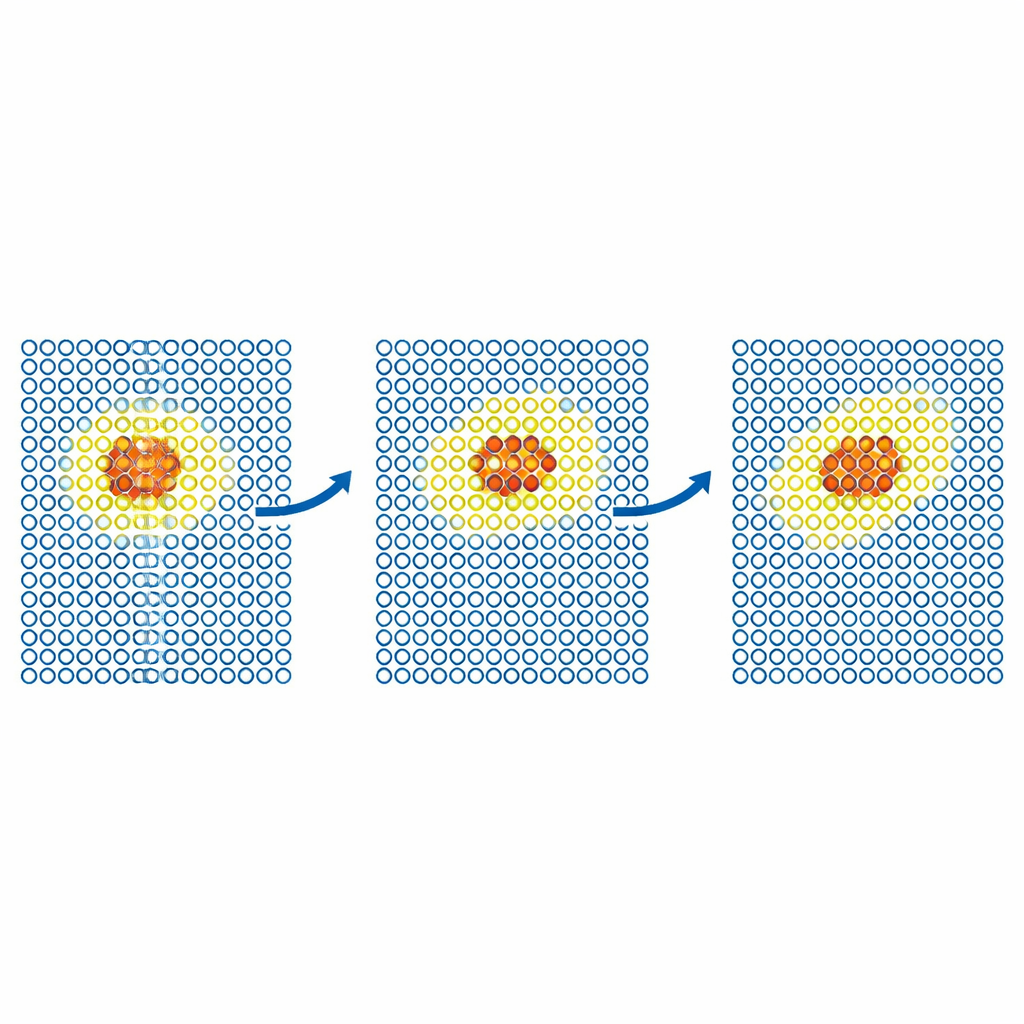
Comparando con experimentos y perspectivas
El estudio de implantación de helio muestra el poder de este enfoque de forma más clara. Al mezclar un modelo de vanguardia aprendido por máquina para las interacciones helio–tungsteno con un potencial más rápido para tungsteno puro, los autores simulan muchos más eventos de impacto y muestras más grandes de lo que sería factible de otro modo, todo en procesadores gráficos. La fracción predicha de átomos de helio que rebotan en la superficie frente a los que se implantan dentro del metal concuerda con mediciones experimentales hasta energías incidentes de aproximadamente 80 electron‑voltios, algo que simulaciones anteriores tuvieron dificultades para alcanzar. Aunque el esquema de mezcla no conserva estrictamente la energía y requiere termostatos suaves, la deriva resultante es pequeña y manejable. En conjunto, ML‑MIX demuestra que combinar cuidadosamente modelos atómicos detallados y simplificados puede superar barreras históricas entre precisión y escala, abriendo la puerta a simulaciones de alta fidelidad y uso rutinario de materiales complejos en entornos realistas.
Cita: Birks, F., Nutter, M., Swinburne, T.D. et al. Efficient and accurate spatial mixing of machine learned interatomic potentials for materials science. npj Comput Mater 12, 110 (2026). https://doi.org/10.1038/s41524-026-01982-6
Palabras clave: potenciales interatómicos aprendidos por máquina, simulación multiescala de materiales, implantación de helio en tungsteno, defectos y dislocaciones, aceleración de dinámica molecular