Clear Sky Science · es
Expansión de clústeres atómicos en grafo para potenciales interatómicos fundacionales de aprendizaje automático
Enseñar a los ordenadores a sentir los átomos
Diseñar nuevos materiales para baterías, aviones o reactores de fusión suele reducirse a una pregunta simple: ¿cómo se empujan y atraen los átomos entre sí? Calcular estas fuerzas exactamente es tan costoso que puede llevar días en un superordenador para un solo material. Este artículo presenta una nueva familia de modelos de aprendizaje automático, llamados GRACE, que actúan como una “calculadora” universal para las fuerzas atómicas a lo largo de gran parte de la tabla periódica. Su objetivo es convertir las simulaciones precisas de materiales complejos en algo rutinario en lugar de heróico.
Un solo modelo para muchos materiales
La mayoría de los campos de fuerzas de aprendizaje automático existentes son herramientas especializadas: funcionan muy bien para unos pocos elementos o compuestos, pero deben reconstruirse desde cero cuando se añaden nuevos elementos. GRACE toma una ruta distinta. Está diseñado desde el principio como un modelo fundacional que puede manejar 89 elementos químicos y una enorme variedad de arreglos atómicos con un único conjunto compartido de reglas. Para ello, los autores se basan en un marco matemático llamado expansión de clústeres atómicos y lo extienden a estructuras de tipo grafo, permitiendo que el modelo describa tanto vecindarios locales de átomos como patrones más extendidos de manera unificada. En lugar de codificar rígidamente cada interacción posible, GRACE aprende “embeddings” compactos que capturan similitudes entre elementos, de modo que el conocimiento sobre un material puede ayudar a describir otro.
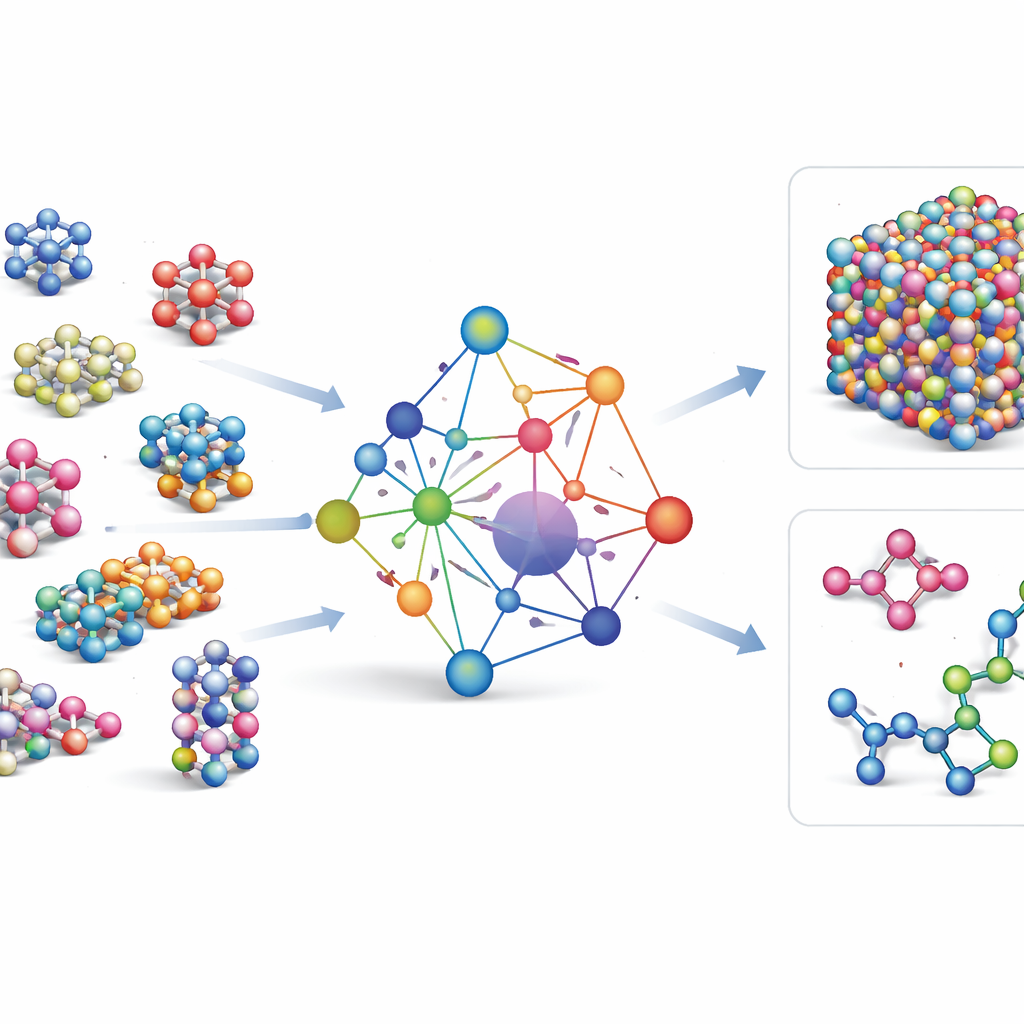
Entrenamiento con un mar de datos atómicos
Para enseñar a GRACE cómo se comportan los átomos, los autores reunieron algunas de las mayores bases de datos públicas de cálculos cuántico-mecánicos. El núcleo es la colección OMat24, que contiene alrededor de 110 millones de simulaciones de materiales inorgánicos, complementada por otras dos que rastrean cómo las estructuras se relajan y evolucionan. En conjunto, estos conjuntos de datos cubren cristales cerca del equilibrio, estructuras deformadas y distorsionadas, líquidos a alta temperatura y más, a lo largo del mismo amplio conjunto de elementos. Los modelos GRACE vienen en varios tamaños, desde versiones más sencillas de una sola capa que sólo observan entornos atómicos locales hasta versiones más profundas de dos capas que efectivamente transmiten “mensajes” entre regiones vecinas. El entrenamiento inicial busca un buen equilibrio entre energías, fuerzas y tensiones internas, y un ajuste posterior afina los modelos para que sean compatibles con bases de referencia ampliamente utilizadas en ciencia de materiales.
Poniendo el modelo a prueba
Un modelo universal solo es útil si rinde de forma fiable en muchas tareas. Por eso los autores someten a GRACE a una exigente batería de pruebas que refleja cómo los científicos usan realmente las simulaciones atomísticas. En un benchmark comunitario para descubrir estructuras cristalinas estables, GRACE se sitúa de forma consistente en el “frente de Pareto”: para una precisión dada, es más rápido que modelos competidores, y para una velocidad dada, es más preciso. Ventajas similares aparecen al predecir la conductividad térmica, una propiedad que depende de manera sensible de pequeños cambios en el movimiento atómico. GRACE también obtiene buenos resultados en propiedades elásticas, energías de superficie, energías de límites de grano y energías de formación de defectos puntuales en muchos metales puros, todas las cuales examinan cómo responden los materiales al estiramiento, corte o daño local. Una larga simulación de dinámica molecular de una sal fundida caliente muestra que el modelo se mantiene numéricamente estable durante nanosegundos mientras reproduce patrones estructurales detallados y tasas de difusión atómica.
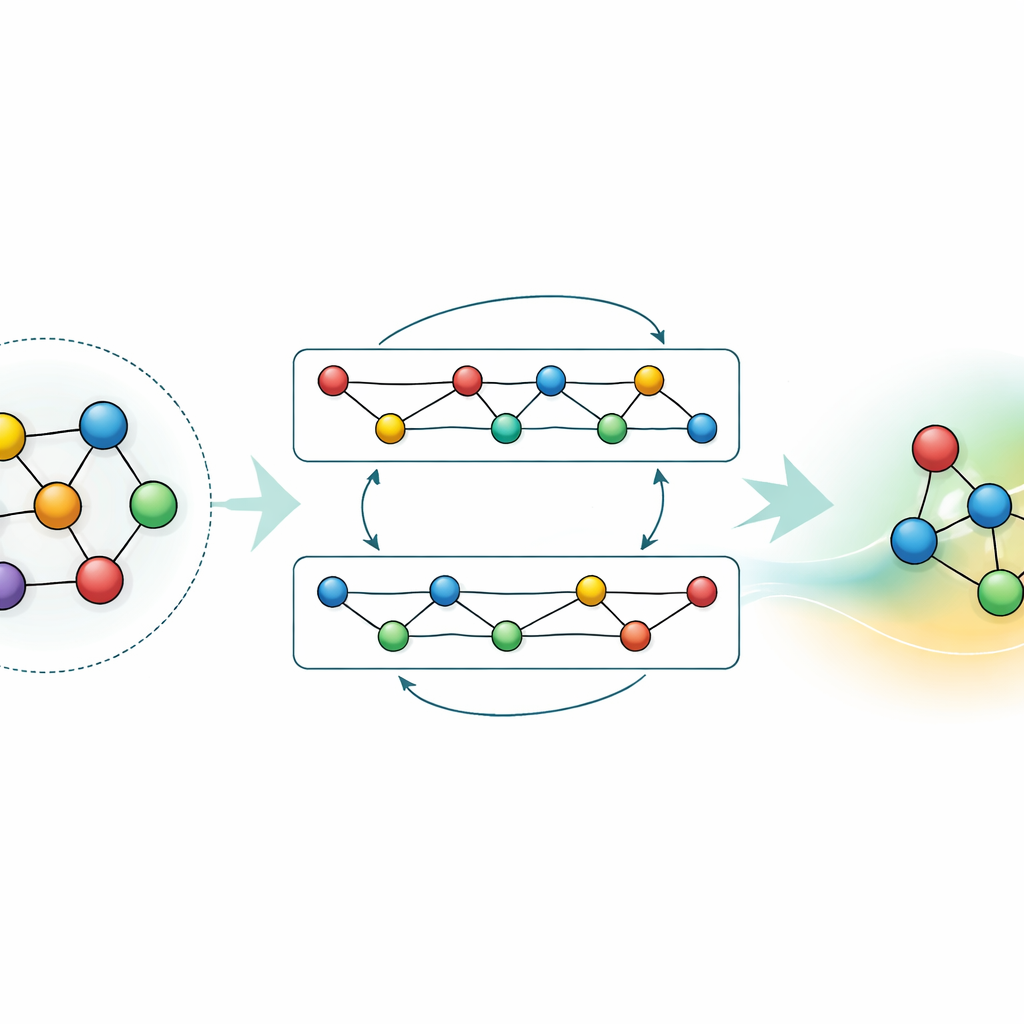
Adaptar y comprimir el conocimiento
Aunque un modelo de propósito general es potente, muchas aplicaciones necesitan bien mayor precisión para un material específico, bien cálculos más rápidos en hardware modesto. Los autores demuestran dos estrategias para lograr esto sin desperdiciar lo que GRACE ya ha aprendido. Primero, afinan el modelo fundacional con conjuntos de datos focalizados, como aleaciones de aluminio-litio o rutas detalladas de combustión del hidrógeno. Para las aleaciones, incluso datos adicionales modestos afinan significativamente las predicciones, superando a modelos entrenados desde cero con la misma información. Para la combustión, un ajuste ingenuo normalmente haría que el modelo “olvidara” lo que sabía sobre otros materiales; al congelar cuidadosamente partes de la red y actualizar sólo parámetros seleccionados, los autores limitan este olvido catastrófico mientras aún ganan precisión para la nueva química. Segundo, muestran cómo destilar el modelo grande en un “estudiante” mucho más sencillo que imita al maestro en sistemas clave. Esta versión destilada se ejecuta alrededor de setenta veces más rápido en CPU y conserva la mayor parte de la precisión, especialmente cuando se entrena con una mezcla de aleaciones complejas y estructuras de referencia más simples etiquetadas por el GRACE original.
Qué significa esto para el diseño futuro de materiales
Este trabajo posiciona a GRACE como una base flexible para la próxima generación de modelado atomístico. En lugar de crear un nuevo potencial para cada material o propiedad, los investigadores pueden partir de un modelo GRACE universal y luego afinarlo o destilarlo según sus necesidades, ahorrando enormes cantidades de tiempo de cálculo y trabajo experto. Los benchmarks muestran que este enfoque no solo iguala a las herramientas existentes; a menudo las supera tanto en velocidad como en fiabilidad, particularmente para propiedades exigentes como el transporte térmico. Para los no especialistas, el mensaje clave es que un único modelo de aprendizaje automático bien diseñado puede ahora actuar como un “motor” ampliamente confiable para experimentos virtuales a lo largo de gran parte de la tabla periódica, acelerando la búsqueda de mejores baterías, catalizadores, aleaciones estructurales y materiales energéticos.
Cita: Lysogorskiy, Y., Bochkarev, A. & Drautz, R. Graph atomic cluster expansion for foundational machine learning interatomic potentials. npj Comput Mater 12, 114 (2026). https://doi.org/10.1038/s41524-026-01979-1
Palabras clave: potenciales interatómicos de aprendizaje automático, modelado de materiales, simulaciones atómicas, modelos fundacionales, expansión de clústeres atómicos en grafo