Clear Sky Science · es
Flujo de trabajo automatizado asistido por potenciales de aprendizaje automático autooptimizantes para el diseño de materiales complejos altamente eficiente
Búsquedas más inteligentes de nuevos materiales
Diseñar nuevos materiales es algo parecido a buscar una aguja en un pajar casi infinito. Desde baterías mejores y ordenadores más rápidos hasta láseres más eficientes y posibles superconductores a temperatura ambiente, muchas tecnologías futuras dependen de descubrir las disposiciones atómicas exactas. Este artículo presenta una forma de dejar que la inteligencia artificial haga la mayor parte de esa búsqueda de forma automática, reduciendo drásticamente el tiempo y el coste necesarios para encontrar compuestos prometedores.
Por qué el rompecabezas de los materiales es tan difícil
Las propiedades de un sólido —qué tan bien conduce la electricidad, cuán resistente es, cómo responde a la luz— vienen determinadas por cómo se disponen sus átomos en patrones tridimensionales llamados estructuras cristalinas. En teoría, se puede usar la mecánica cuántica para calcular qué disposiciones son estables y cuáles serán sus propiedades. En la práctica, estos cálculos cuánticos son tan exigentes que solo una fracción mínima de todos los materiales posibles puede evaluarse. El desafío crece rápidamente cuando intervienen más de dos elementos químicos, porque el número de combinaciones y disposiciones atómicas se dispara, haciendo imposible una búsqueda a ciegas.
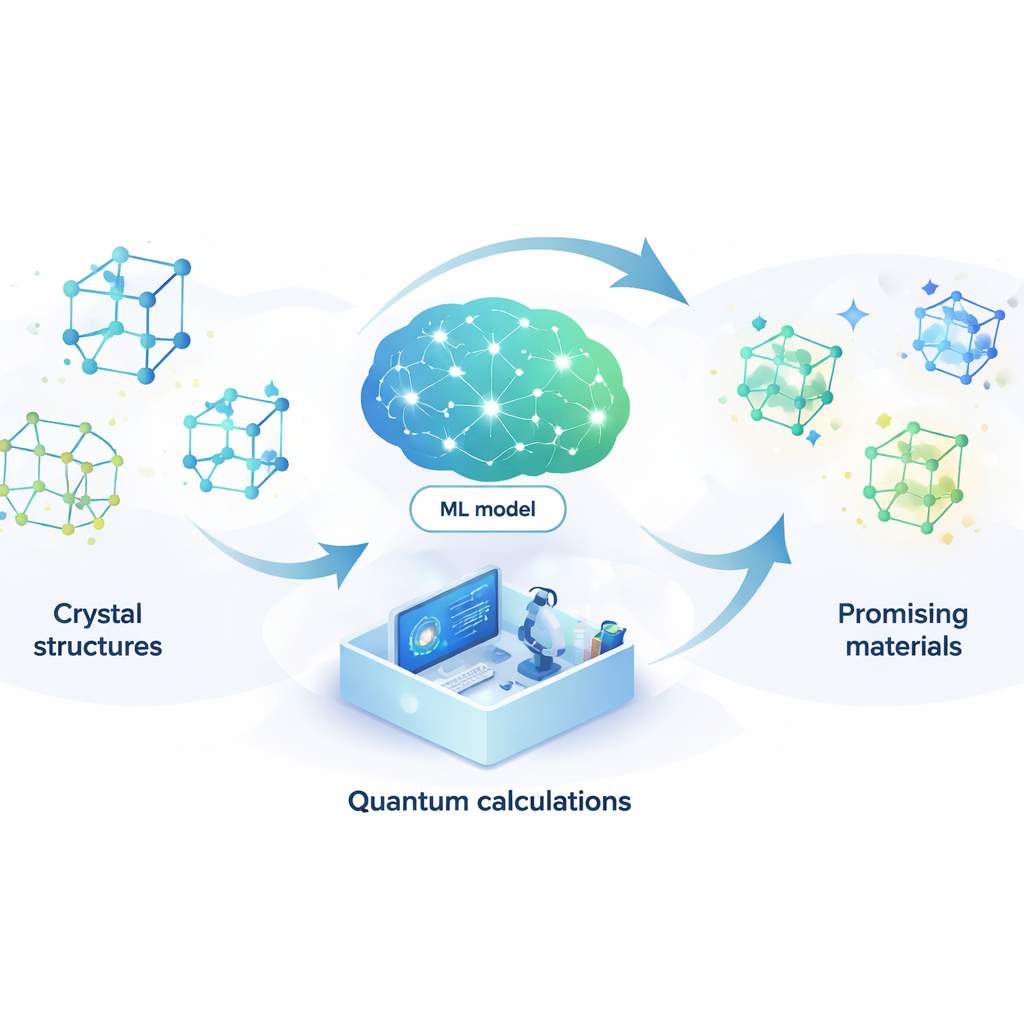
Dejar que un modelo aprendiente sustituya a la física cuántica
Para abordar este problema, los autores construyen un modelo de aprendizaje automático que puede imitar los resultados de costosos cálculos cuánticos a una fracción mínima del coste. Su modelo, llamado red neuronal acoplada por atención (ACNN), aprende cómo la energía de un material depende de las posiciones y los tipos de sus átomos. Una vez entrenado, puede estimar muy rápidamente si una estructura cristalina propuesta es probable que sea estable y qué fuerzas actúan sobre cada átomo. De forma crucial, el modelo está diseñado para respetar requisitos físicos básicos, como que desplazar o rotar todo el cristal no debe cambiar su energía total.
Un bucle de descubrimiento de materiales que se mejora a sí mismo
En lugar de entrenar el modelo una sola vez con la esperanza de que funcione en todas partes, los autores lo integran en un bucle autooptimizante. El proceso empieza con un pequeño conjunto de estructuras cristalinas aleatorias, que se evalúan con cálculos cuántico-mecánicos completos y se usan para entrenar una ACNN inicial. Este modelo se utiliza luego para relajar millones de estructuras de prueba, encontrando rápidamente mínimos locales de energía: fases candidatas estables o casi estables. El flujo de trabajo marca automáticamente dos tipos de estructuras especialmente valiosas: las que parecen muy estables y las que parecen no físicas o sospechosas. Solo estos casos seleccionados se envían de vuelta al solucionador cuántico costoso, y los nuevos resultados se incorporan al modelo para volver a entrenarlo. A lo largo de muchas rondas, el modelo se vuelve progresivamente más preciso en las regiones del espacio de estructuras que más importan.
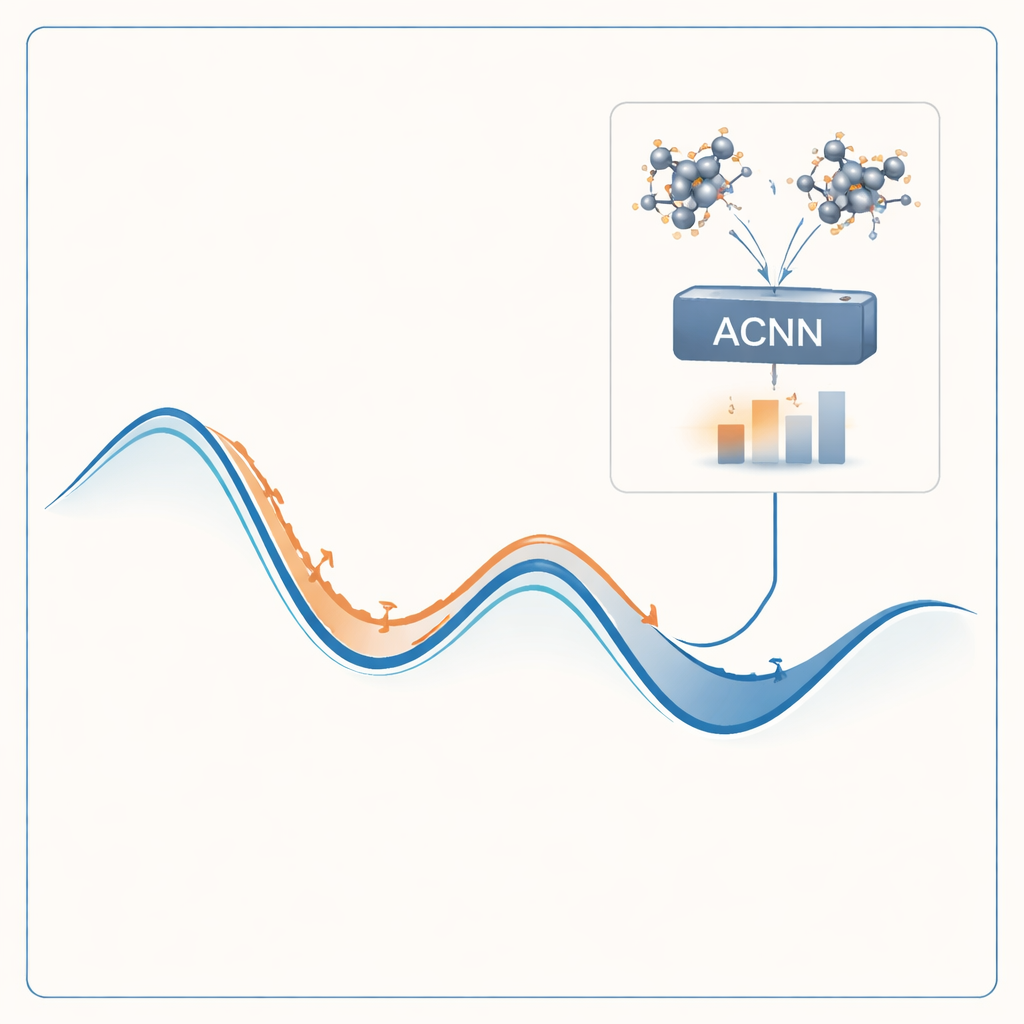
Poner el método a prueba
El equipo demuestra su enfoque en dos sistemas exigentes. El primero es una mezcla a alta presión de magnesio, calcio e hidrógeno, una familia de compuestos de gran interés para la superconductividad a alta temperatura. Al explorar casi seis millones de estructuras de prueba, su flujo de trabajo descubre una nueva fase estable, MgCa₃H₂₃, y varias estructuras relacionadas ricas en hidrógeno en forma de “jaula”. Los cálculos sugieren que algunas de estas podrían ser superconductoras a temperaturas superiores al punto de ebullición del nitrógeno líquido bajo presiones extremas. La segunda prueba se dirige a un sistema de cuatro elementos que contiene berilio, fósforo, nitrógeno y oxígeno, elegido por su potencial para albergar cristales que conviertan de forma eficiente la luz láser en longitudes de onda ultravioleta profundas. Aquí el método relaja más de nueve millones de estructuras e identifica tres fases termodinámicamente estables con bandas prohibidas muy amplias y características ópticas prometedoras.
De la fuerza bruta al descubrimiento guiado
En ambos ejemplos, el flujo de trabajo automatizado consigue aceleraciones del orden de diez mil veces en comparación con el uso exclusivo de cálculos cuánticos, mientras sigue localizando de forma fiable estructuras que merecen un estudio más detallado. Para un público no especialista, el mensaje clave es que gran parte del descubrimiento de materiales puede ahora ser manejado por un sistema de aprendizaje que aprende dónde tiene incertidumbre y pide cálculos de alta precisión dirigidos solo cuando es necesario. Este tipo de búsqueda autocorrectiva asistida por IA abre la puerta a explorar mezclas de elementos mucho más complejas de lo que antes era factible, aumentando las posibilidades de descubrir nuevos superconductores, cristales ópticos y otros materiales funcionales que sustentan las tecnologías de próxima generación.
Cita: Li, J., Feng, J., Luo, J. et al. Self-optimizing machine learning potential assisted automated workflow for highly efficient complex systems material design. npj Comput Mater 12, 101 (2026). https://doi.org/10.1038/s41524-026-01971-9
Palabras clave: descubrimiento de materiales, potenciales de aprendizaje automático, predicción de estructuras cristalinas, hidruros superconductores, cristales ópticos no lineales