Clear Sky Science · es
Cálculo desde primeros principios de estructuras de dislocación y transformaciones de fase impulsadas por esfuerzo en óxidos estratificados para baterías de iones de sodio
Por qué los defectos diminutos importan para las baterías del futuro
Mientras el mundo mira más allá del litio hacia baterías de iones de sodio, más baratas y con mayor abundancia, un mundo oculto dentro de los materiales del cátodo se vuelve crucial: defectos cristalinos diminutos llamados dislocaciones. Estas irregularidades lineales, del tamaño de unos pocos átomos, permiten que el material se adapte cuando los iones de sodio entran y salen, pero también pueden desencadenar el daño estructural que acorta la vida útil de la batería. Este trabajo usa simulaciones computacionales a nivel cuántico para desvelar cómo se forman, se mueven y promueven cambios de fase las dislocaciones en cátodos estratificados de sodio, ofreciendo orientaciones para diseñar baterías más duraderas y robustas.
Capas atómicas apiladas que deben mantener su forma
Muchos cátodos prometedores para iones de sodio se construyen a partir de pilas de láminas atómicas planas. Los iones de sodio se sitúan entre capas de metal de transición y oxígeno en una disposición ordenada “O3” cuando están completamente sodiadas, pero al cargar y descargar repetidamente la estructura tiende a desplazarse hacia otro patrón de apilamiento, denominado “P3”. Estos cambios en el alineamiento de las capas —la secuencia de apilamiento— pueden ser reversibles e inofensivos, o bien pueden provocar colapso, fisuras y pérdida de capacidad. Los autores se centran en una familia de óxidos estratificados, Na(TM)O₂ con TM = Ti, Cr, Mn, Fe, Co o Ni, y se preguntan: ¿qué tan fácil es que estos materiales reorganicen su apilamiento y qué papel juegan las dislocaciones en esos procesos? 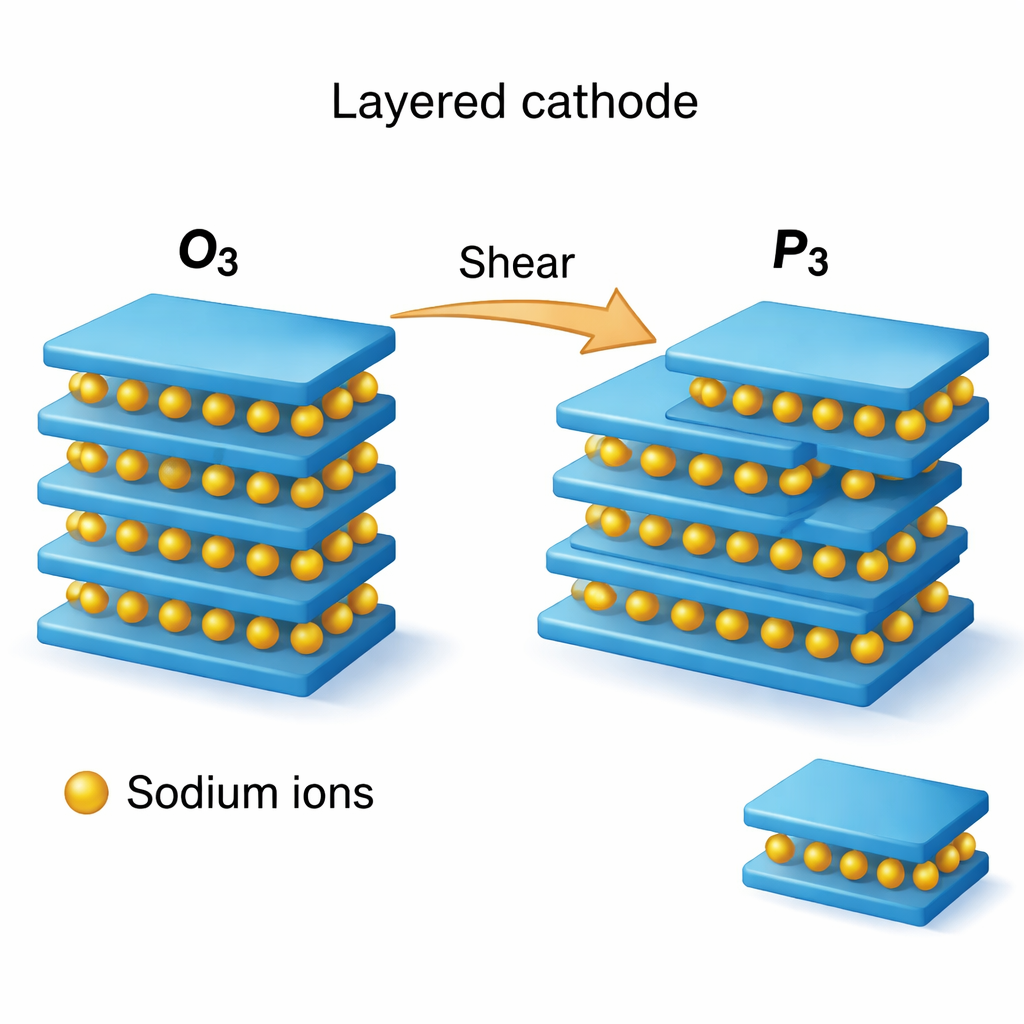
Cartografiar cómo prefieren deslizarse las capas
Para responder a esto, los investigadores calculan primero las denominadas superficies de energía de fallas de apilamiento generalizadas. En términos simples, toman dos mitades del cristal, deslizan una mitad sobre la otra en distintas direcciones y calculan cuánto cuesta energéticamente cada desplazamiento. Los caminos de baja energía en este mapa revelan cómo prefieren deslizarse las capas y si es probable que se formen estados “fallados” intermedios —reordenamientos locales del apilamiento. En todos los compuestos estudiados encuentran que un estado fallado de tipo P3 es posible, pero resulta especialmente favorecido en materiales a base de cobalto y níquel, que muestran profundos mínimos de energía para esta configuración. En contraste, un apilamiento más drástico de tipo O1 no aparece como un estado estable bajo las condiciones que modelan, lo que sugiere que los cambios más suaves O3↔P3 son intrínsecamente más accesibles.
Cómo se ven las dislocaciones dentro de estos cátodos
Los cristales reales no se cortan como bloques rígidos perfectos; se deforman mediante el movimiento de dislocaciones. Empleando un modelo semi‑discreto de Peierls–Nabarro informado por sus datos cuántico‑mecánicos, los autores reconstruyen la estructura interna —o “núcleo”— tanto de dislocaciones de borde como de tornillo en el plano de deslizamiento clave paralelo a las capas. Encuentran que los núcleos de las dislocaciones son muy estrechos, de solo unos pocos nanómetros de ancho, lo que confirma que estos materiales son mecánicamente rígidos. Las dislocaciones de borde tienden a dividirse en dos dislocaciones “parciales” separadas por una delgada franja que localmente presenta apilamiento de tipo P3, especialmente en los óxidos ricos en Co y Ni donde el estado P3 es energéticamente favorecido. Las dislocaciones de tornillo suelen permanecer más compactas, pero en algunas composiciones (de nuevo, notablemente Co y Ni) también pueden dividirse y crear regiones estrechas similares a P3.
Con qué facilidad se mueven los defectos bajo los esfuerzos de la batería
A continuación, el estudio estima la tensión de Peierls —la tensión cortante mínima necesaria para iniciar el movimiento de una dislocación a través de la red. Esta magnitud actúa como una resistencia a escala microscópica para defectos individuales. Para todos los materiales examinados, las tensiones requeridas (de unos pocos a unas pocas decenas de megapascales) se encuentran dentro del rango de tensiones esperadas cuando se insertan y extraen iones de sodio durante los ciclos. Eso significa que el movimiento de dislocaciones no solo es posible, sino probable en condiciones de operación realistas. Los cálculos también muestran que algunas estructuras, en particular variantes monoclínicas de óxidos de Mn y Ni, ofrecen mayor resistencia a ciertos tipos de movimiento de dislocaciones porque sus caminos preferidos de deslizamiento de baja energía son más restringidos. 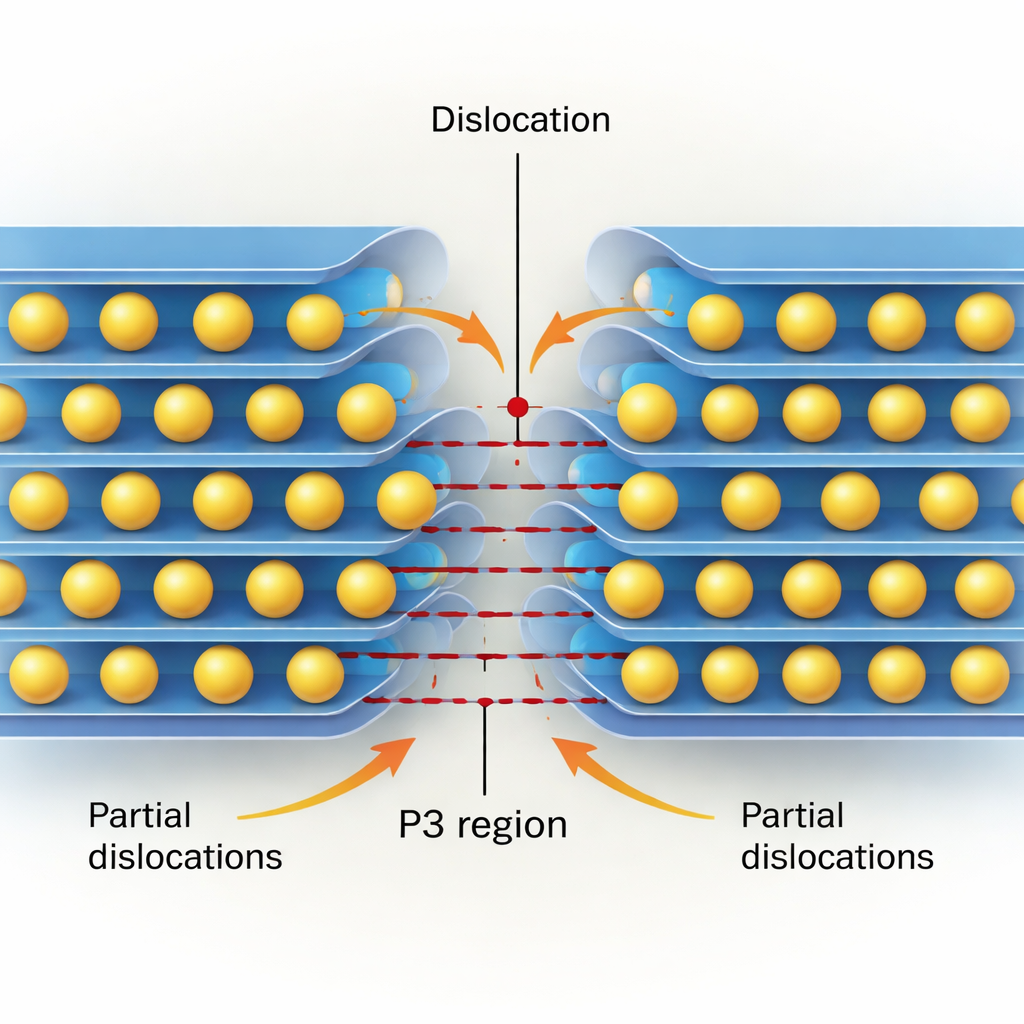
Las dislocaciones como motores del cambio de fase
Uniendo estas piezas, los autores proponen un panorama en el que las dislocaciones impulsan activamente la transformación de fase O3→P3. En un cátodo totalmente sodiado, las dislocaciones existentes o de nueva formación pueden dividirse en parciales, sembrando pequeñas regiones similares a P3 a lo largo de su línea. A medida que se extrae sodio, el paisaje energético local cambia de modo que la configuración P3 se vuelve cada vez más estable. La franja P3 entre las dislocaciones parciales entonces se ensancha, y los iones de sodio saltan hacia los nuevos sitios prismáticos, permitiendo que la región P3 crezca y se propague por la partícula. Tras muchos ciclos, la acumulación y el movimiento de estos defectos también puede contribuir a microgrietas y fases irreversibles, conectando procesos a escala atómica directamente con la degradación de la batería.
Reglas de diseño para baterías de sodio más resistentes
Para un público no especialista, el mensaje clave es que la vida útil de las baterías de iones de sodio depende no solo de qué elementos se eligen, sino también de cómo prefieren deslizarse sus capas atómicas y de la facilidad con que se mueven las dislocaciones. Al cartografiar estos comportamientos desde primeros principios, el estudio ofrece pautas de diseño: las químicas que mantienen las energías de falla de apilamiento poco profundas y controlan el movimiento de dislocaciones pueden favorecer transiciones O3↔P3 suaves y reversibles y resistir la formación de grietas. En términos prácticos, eso significa que los ingenieros pueden ajustar la composición y la estructura para gestionar estos defectos diminutos, allanando el camino hacia baterías de iones de sodio más baratas que las celdas de litio actuales y lo bastante duraderas para el almacenamiento de energía a gran escala.
Cita: Arcelus, O., Carrasco, J. First-principles computation of dislocation structures and stress-driven phase transformations in layered oxides for Na-ion batteries. npj Comput Mater 12, 96 (2026). https://doi.org/10.1038/s41524-026-01965-7
Palabras clave: baterías de iones de sodio, cátodos estratificados, dislocaciones, transformaciones de fase, degradación de materiales