Clear Sky Science · es
Aprendizaje de dinámica molecular coarse-grained eficiente en datos a partir de fuerzas y ruido
Por qué importa reducir las moléculas
Simular el movimiento incesante de cada átomo en una proteína y el agua que la rodea es una de nuestras mejores herramientas para entender cómo funciona la vida a escala molecular. Pero estas simulaciones a nivel atómico son tan exigentes computacionalmente que seguir a una proteína mientras se pliega, despliega o interactúa con otras durante tiempos relevantes biológicamente puede llevar meses en un superordenador. Este artículo presenta una nueva forma de construir modelos rápidos y simplificados de proteínas que siguen comportándose como sus contrapartes atómicas completas, necesitando al mismo tiempo mucha menos información de entrenamiento y potencia de cálculo que antes.
De cada átomo a una imagen más simple
La dinámica molecular tradicional rastrea cada átomo y calcula las fuerzas entre ellos en cada diminuto paso temporal. Para acelerar el proceso, los científicos usan a menudo modelos coarse-grained, que agrupan muchos átomos en un número menor de “perlas”. Estos modelos reducidos se ejecutan mucho más rápido, pero históricamente han tenido dificultades para igualar la precisión de las simulaciones atomísticas completas, especialmente en proteínas con comportamientos de plegamiento complejos. Trabajos recientes se han orientado hacia el aprendizaje automático para descubrir automáticamente mejores campos de fuerza coarse-grained, pero entrenar estos modelos típicamente ha requerido millones de instantáneas detalladas, cada una etiquetada con las fuerzas sobre cada átomo: una enorme carga de datos y cálculo.
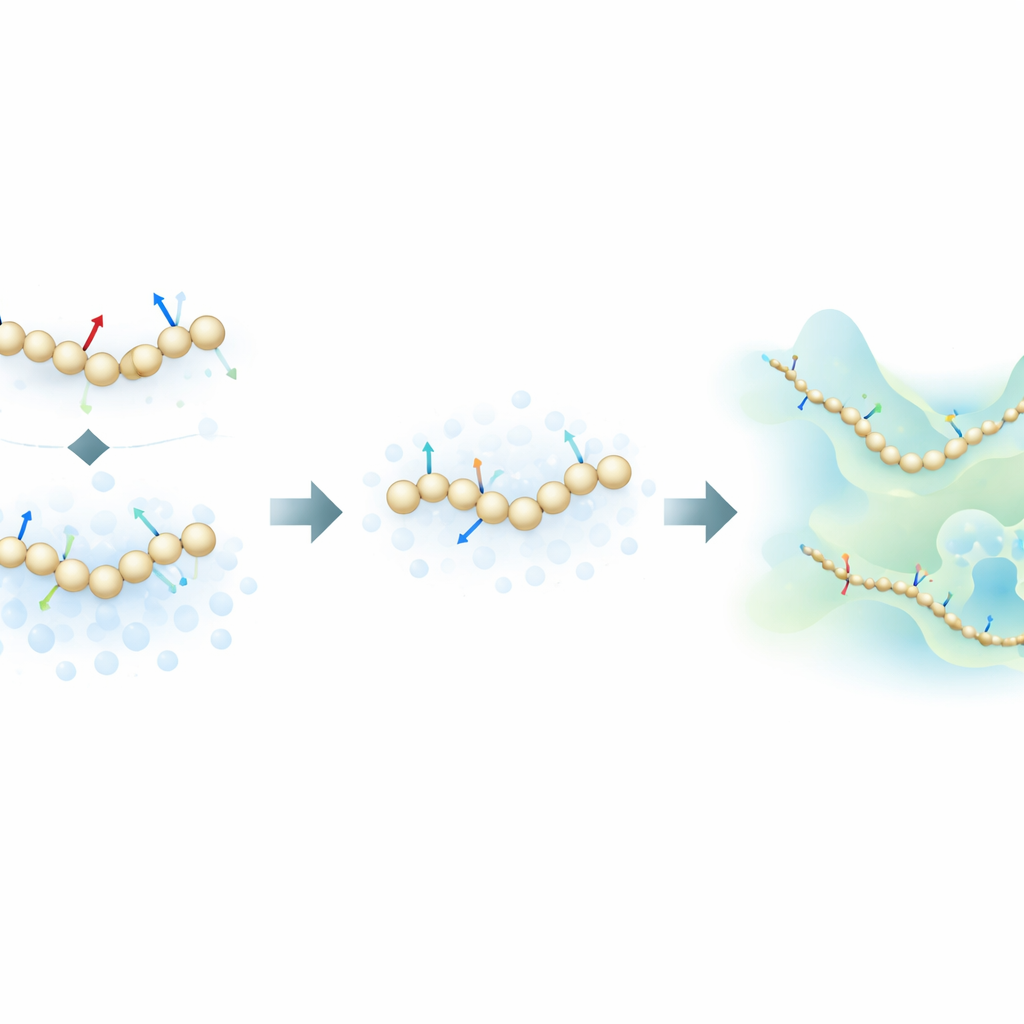
Mezclando fuerzas físicas con ruido informativo
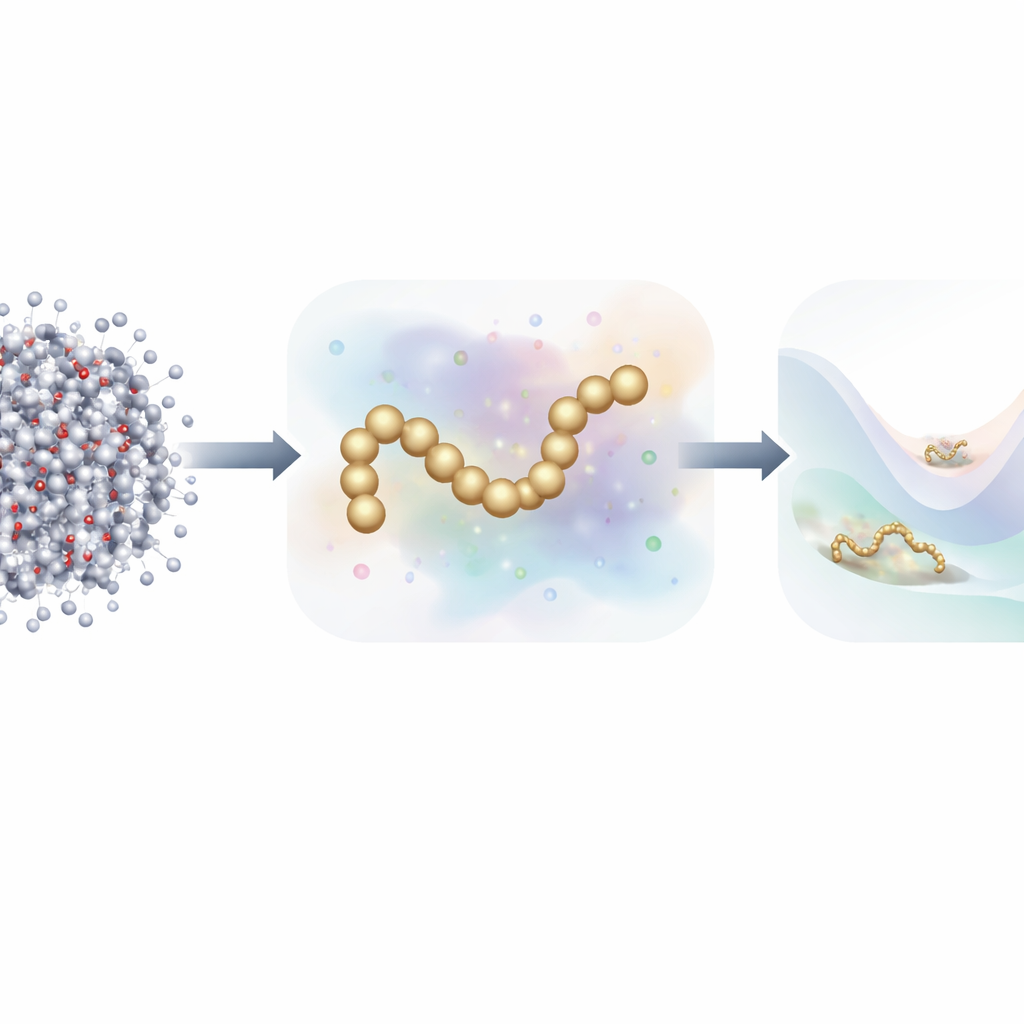
Los autores proponen una estrategia de entrenamiento novedosa que toma inspiración de los modelos generativos de difusión—la misma clase de algoritmos detrás de muchos generadores de imágenes de IA modernos. En lugar de aprender solo a partir de las fuerzas físicas calculadas en simulaciones atomísticas, su método también aprende a partir de cómo se distribuyen las estructuras moleculares en el espacio al añadir deliberadamente ruido controlado a las configuraciones coarse-grained. En este marco, el ruido no es solo una molestia a eliminar; se convierte en una fuente adicional de información. Al unificar matemáticamente el enfoque tradicional de “emparejamiento de fuerzas” con técnicas de eliminación de ruido de los modelos de difusión, el método puede inferir el paisaje energético subyacente de una proteína usando muchas menos muestras etiquetadas.
Enseñar a modelos simples a imitar proteínas complejas
Para probar su idea, los investigadores entrenaron modelos coarse-grained basados en redes neuronales para varias proteínas de complejidad creciente: las mini proteínas pequeñas Chignolin y Trp-Cage, la algo mayor NTL9, y la proteína de 76 residuos Ubiquitina. Compararon tres modos de entrenamiento: usando solo fuerzas atomísticas, usando solo información basada en ruido, y combinando ambos. Para las proteínas más pequeñas, demostraron que el nuevo enfoque combinado puede reproducir las características clave del paisaje de plegamiento—como la estabilidad relativa de los estados plegado y desplegado y la presencia de intermedios—usando hasta cien veces menos datos de entrenamiento que los métodos estándar de emparejamiento de fuerzas. Sorprendentemente, en regímenes con pocos datos, incluso los modelos entrenados únicamente con información basada en ruido a menudo igualaron o superaron la precisión del entrenamiento solo con fuerzas.
Alcanzando sistemas proteicos más grandes y difíciles
Ubiquitina es una prueba más exigente: capturar su plegamiento y despliegue a temperaturas realistas ha requerido históricamente hardware especializado y ejecuciones atomísticas extremadamente largas. Aquí, los autores entrenan modelos coarse-grained usando un conjunto de datos modesto consistente en simulaciones de equilibrio cortas alrededor del estado plegado más simulaciones no equilibrio “tiradas” que estiran forzosamente la proteína. A pesar de este conjunto de entrenamiento sesgado y de la ausencia de una referencia atomística perfecta en las mismas condiciones, el modelo entrenado con fuerzas y ruido recupera una imagen realista en la que los estados plegado y desplegado coexisten, con el estado plegado favorecido en estabilidad. En contraste, un modelo entrenado solo con fuerzas no logra estabilizar el estado plegado en absoluto, mientras que un modelo solo con ruido prefiere estructuras desplegadas. Cabe destacar que ninguno de los modelos coarse-grained memoriza simplemente las formas extremadamente estiradas de los datos de entrenamiento, lo que indica que el paisaje energético aprendido tiene sentido físico y no es solo una huella de las trayectorias de entrada.
Qué significa esto para las simulaciones futuras
Al convertir el ruido en una señal de entrenamiento y fusionarlo con las fuerzas físicas, este trabajo muestra que se pueden construir modelos coarse-grained precisos de proteínas a partir de conjuntos de datos mucho más pequeños y menos ideales de lo que se pensaba. En la práctica, eso significa que los investigadores ya no necesitarán simulaciones atomísticas de milisegundos en supercomputadores especializados antes de poder explorar el comportamiento de un biomolécula con dinámica coarse-grained aprendida por máquina. En su lugar, simulaciones más modestas en hardware ampliamente accesible podrían ser suficientes para entrenar modelos reducidos potentes que capturen las rutas de plegamiento clave y los equilibrios termodinámicos. Aunque quedan preguntas sobre cómo elegir e interpretar mejor el ruido añadido y cómo funcionará el método en ensamblajes biomoleculares aún mayores y más complejos, este enfoque reduce sustancialmente la barrera para usar simulaciones coarse-grained basadas en datos como una herramienta de rutina en la ciencia molecular.
Cita: Durumeric, A.E.P., Chen, Y., Pasos-Trejo, A.S. et al. Learning data-efficient coarse-grained molecular dynamics from forces and noise. Nat Commun 17, 2493 (2026). https://doi.org/10.1038/s41467-026-70818-0
Palabras clave: dynamics molecular coarse-grained, campos de fuerza por aprendizaje automático, simulaciones de plegamiento de proteínas, modelos de difusión en química, simulación eficiente en datos