Clear Sky Science · es
Una mutación patógena de Tau provoca disfunción autófago‑lisosomal que limita la degradación de Tau en un modelo de demencia frontotemporal
Cuando los equipos de limpieza del cerebro se quedan atrás
¿Por qué algunas personas desarrollan problemas devastadores de memoria y conducta décadas antes de la vejez? Este estudio aborda esa pregunta al centrarse en una sola proteína cerebral, la Tau, y en los diminutos “centros de reciclaje” celulares que normalmente la mantienen bajo control. Observando neuronas humanas vivas con microscopios de altísima resolución, los investigadores muestran cómo una mutación patógena de Tau obstruye el sistema de eliminación de desechos de la célula y cómo estimular ese sistema con una pequeña molécula puede ayudar a despejar el atasco. Sus hallazgos podrían señalar nuevas estrategias terapéuticas para ciertas formas de demencia.
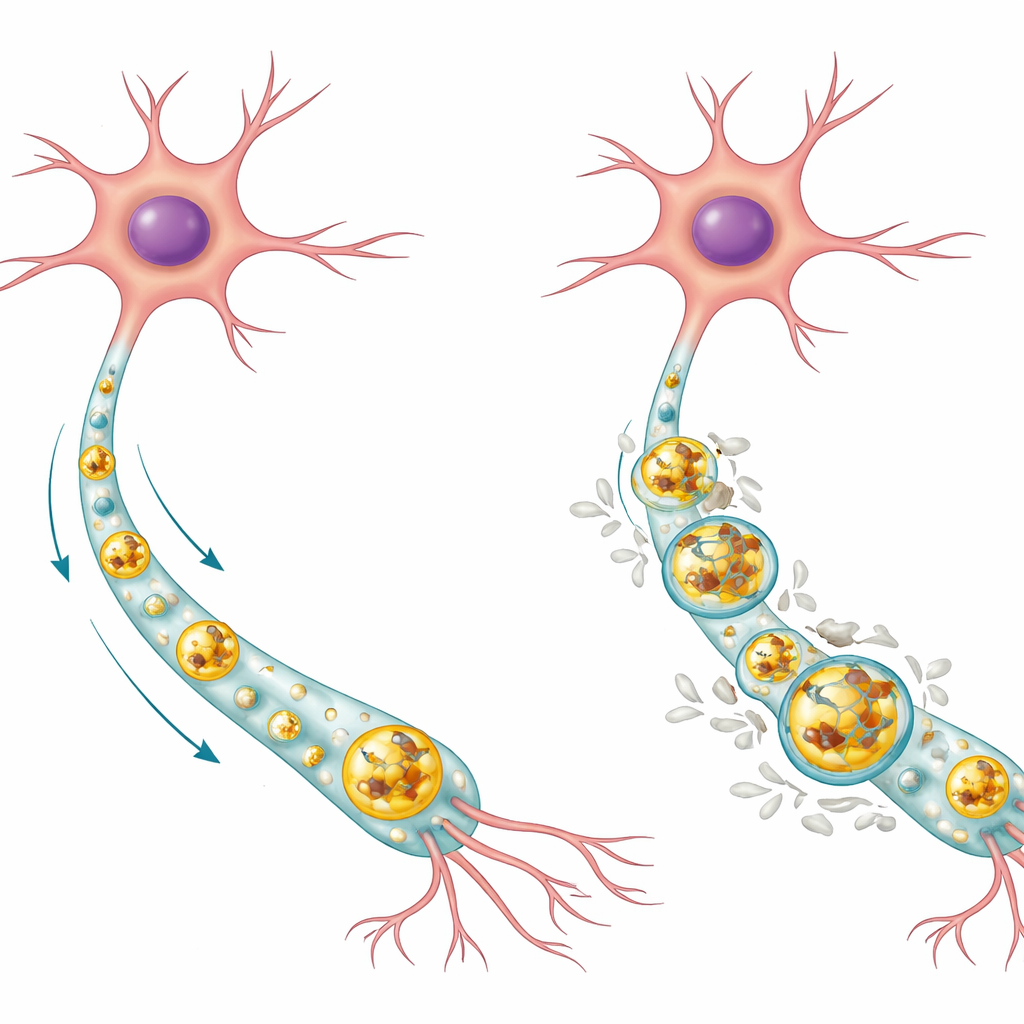
Cómo las neuronas normalmente sacan la basura
Las neuronas son células de larga vida que no pueden simplemente dividirse para diluir material dañado, por lo que dependen en gran medida de sistemas internos de limpieza. Una vía clave es la autófago‑lisosomal. En este proceso, proteínas no deseadas y partes desgastadas se encierran en sacos membranosos llamados autofagosomas, que luego se fusionan con compartimentos llenos de enzimas conocidos como lisosomas, donde la carga se descompone y recicla. En neuronas humanas sanas, los autores hallaron que la proteína Tau normal tiende a acumularse dentro del centro ácido de los lisosomas, donde puede degradarse, mientras que la forma fosforilada de Tau (una modificación química vinculada a la enfermedad) se localiza más en la membrana externa del lisosoma. La mayoría de los lisosomas en células sanas estaban libres de Tau por completo, lo que sugiere que este sistema suele mantener los niveles de Tau bajos y bien controlados.
Qué falla en una forma genética de demencia
El equipo se centró en una mutación del gen MAPT, denominada p.R406W, que provoca una forma hereditaria de demencia frontotemporal y puede imitar la pérdida de memoria al estilo del Alzheimer. Usando tecnología de células madre, reprogramaron células de piel de pacientes en células madre pluripotentes inducidas y posteriormente en grandes cantidades de neuronas humanas que o bien portaban la mutación o habían sido editadas genéticamente para volver a la normalidad. En las neuronas mutantes, tanto la Tau total como la Tau fosforilada eran notablemente más altas, no porque las células produjeran más Tau, sino porque la degradaban con menos eficiencia. Imágenes de súper resolución revelaron que casi todos los lisosomas en las células mutantes estaban repletos de Tau y, especialmente, con Tau fosforilada recubriendo la membrana lisosomal. Esta acumulación señalaba que la principal vía de eliminación de proteínas de la célula estaba atascada.
Centros de reciclaje obstruidos y tráfico lento
Al examinar más de cerca la maquinaria de reciclaje, los investigadores observaron que los lisosomas en neuronas mutantes eran más numerosos, de mayor tamaño y tendían a situarse más lejos del cuerpo celular. La imagen en vivo con tintes fluorescentes mostró que estos lisosomas se movían más despacio y recorrían distancias más cortas a lo largo de las fibras nerviosas, a pesar de que las vías de microtúbulos subyacentes parecían normales. Las neuronas mutantes también contenían más autofagosomas, más de la proteína adaptadora de carga p62 y gotículas lipídicas adicionales, signos de que el material se marcaba para su eliminación pero no se descomponía por completo. Usando un reportero sensible al pH, encontraron que los autofagosomas en células mutantes a menudo no se fusionaban adecuadamente con los lisosomas, lo que conducía a una acumulación de vesículas de reciclaje “a medio terminar” y defectos generalizados en la limpieza celular, no solo para Tau sino también para otras cargas.
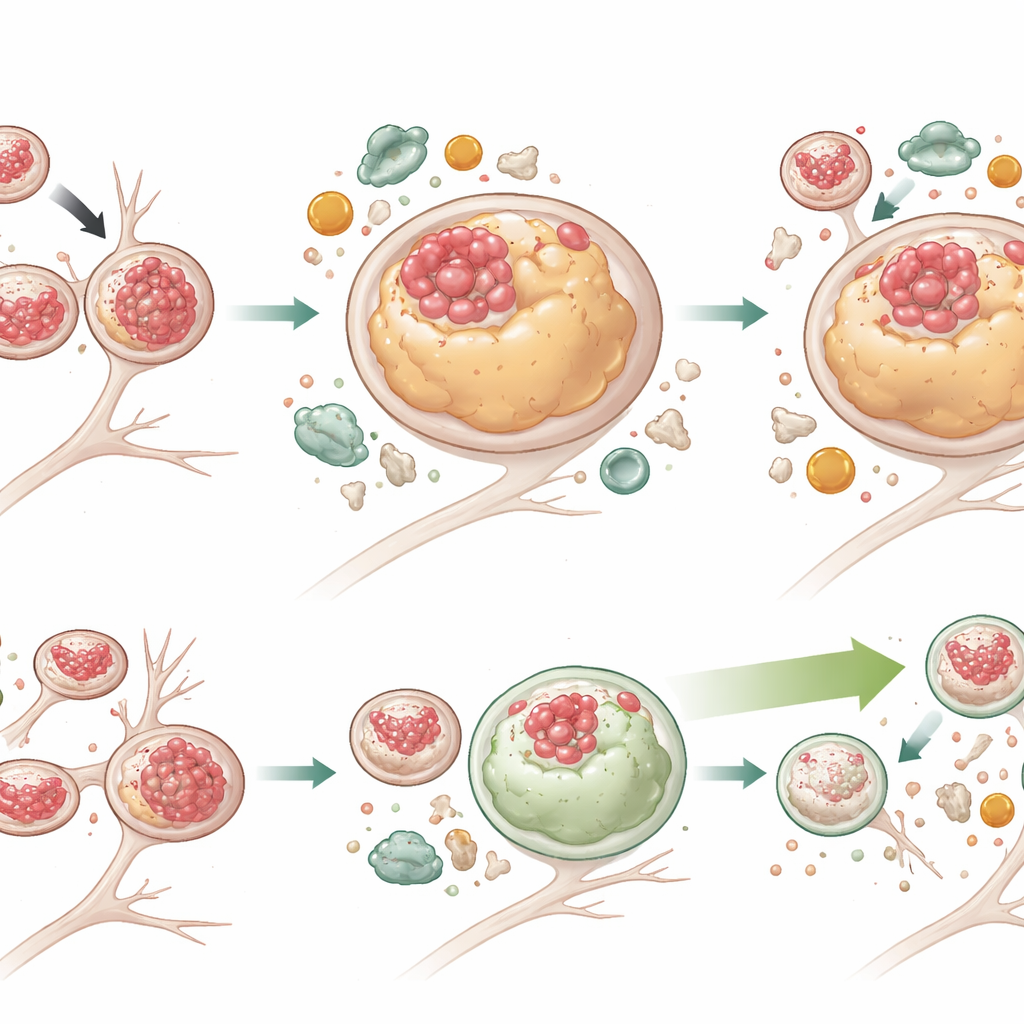
Potenciar la limpieza celular sin arreglar el atasco del tráfico
Para comprobar si mejorar la autofagia podía superar estos problemas, el equipo trató las neuronas con G2‑567, una pequeña molécula previamente demostrada para estimular el sistema autofago‑lisosomal. Tras dos semanas de tratamiento, las neuronas mutantes presentaron niveles sustancialmente menores tanto de Tau total como de Tau fosforilada, y muchos más lisosomas volvieron a estar libres de Tau. Los lisosomas también se redujeron hacia un tamaño más normal. Los marcadores de autofagia activa aumentaron, mientras que p62—un indicador de degradación bloqueada—disminuyó en las células mutantes, demostrando una ruptura de carga más eficaz. Curiosamente, G2‑567 no corrigió todos los defectos: los lisosomas en neuronas mutantes aún tendían a situarse más lejos del cuerpo celular y a moverse con lentitud, y una proteína adaptadora (JIP3) relacionada con el transporte lisosomal se mantuvo elevada. Esto sugiere que las funciones de transporte y degradación de los lisosomas pueden desacoplarse parcialmente, y que mejorar solo la degradación puede ser suficiente para reducir la acumulación tóxica de Tau.
Qué significa esto para futuros tratamientos contra la demencia
Para un lector no especializado, la conclusión clave es que en este modelo genético de demencia frontotemporal, el problema no es únicamente que Tau se vuelva anómala; es que el sistema de reciclaje de la neurona no da abasto. La mutación p.R406W de Tau altera directamente varios pasos de la vía autofago‑lisosomal, provocando que Tau—especialmente su forma fosforilada—se acumule sobre y dentro de los lisosomas, junto con otro material no degradado. Al empujar farmacológicamente la maquinaria de limpieza celular para que trabaje más, los investigadores pudieron disminuir los niveles de Tau y normalizar el tamaño lisosomal, pese a que persistieran defectos de transporte. Estos hallazgos refuerzan la idea de que fármacos diseñados para potenciar de manera segura la autofagia y la función lisosomal podrían ayudar a restaurar el equilibrio proteico en las demencias relacionadas con Tau y quizá en condiciones más comunes, como la enfermedad de Alzheimer.
Cita: Mirfakhar, F.S., Marsh, J.A., Sato, C. et al. A pathogenic Tau mutation drives autophagy-lysosome dysfunction that limits Tau degradation in a model of frontotemporal dementia. Nat Commun 17, 2699 (2026). https://doi.org/10.1038/s41467-026-70473-5
Palabras clave: proteína tau, autofagia, disfunción lisosomal, demencia frontotemporal, neurodegeneración