Clear Sky Science · es
DiNovo permite la secuenciación de péptidos de novo con alta cobertura y alta confianza mediante proteasas espejo y aprendizaje profundo
Ver las proteínas con nuevo detalle
Las proteínas son las pequeñas máquinas que mantienen vivas nuestras células, pero leer completamente sus componentes sigue siendo sorprendentemente difícil. Este artículo presenta DiNovo, un nuevo sistema de software que ayuda a los científicos a “leer” fragmentos proteicos de forma mucho más completa y fiable que antes. Combinando un truco bioquímico ingenioso con la inteligencia artificial moderna, promete descubrir proteínas ocultas, marcadores de enfermedad e incluso dianas inmunitarias que los métodos tradicionales suelen pasar por alto.
Por qué es tan difícil leer fragmentos proteicos
La mayoría del análisis de proteínas hoy en día se basa en cortar las proteínas en piezas más pequeñas, llamadas péptidos, y luego pesar sus fragmentos en un espectrómetro de masas. A partir de estos pesos, los ordenadores intentan reconstruir la secuencia original del péptido, como resolver un crucigrama a partir de pistas parciales. Los métodos existentes suelen asumir que los péptidos proceden de bases de datos de proteínas conocidas, lo que funciona bien para proteínas familiares pero falla con las nuevas o inesperadas. La denominada secuenciación de novo evita esta limitación intentando leer los péptidos directamente a partir de los datos, pero a menudo se queda corta porque faltan algunos fragmentos y porque algunos péptidos nunca se cortan de forma limpia en primer lugar.
Usar enzimas espejo para rellenar los huecos
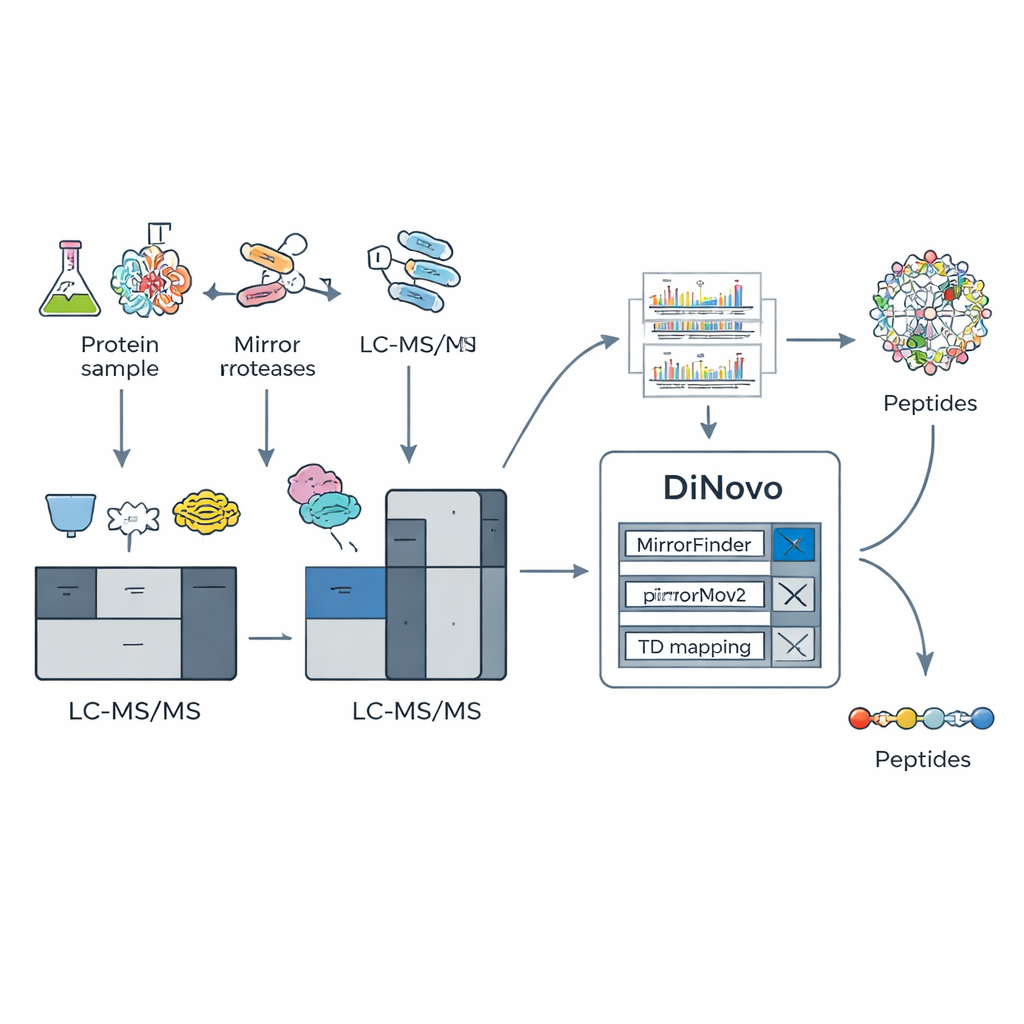
La idea clave detrás de DiNovo es usar pares de “proteasas espejo”: pares de enzimas de corte que fragmentan las proteínas en lados opuestos del mismo tipo de aminoácido. Por ejemplo, una enzima corta justo antes de una lisina, mientras que su pareja corta justo después de esa lisina. Esto produce dos péptidos relacionados que comparten el mismo segmento interno pero tienen extremos distintos. Cuando se analizan estos péptidos “espejo”, sus espectros de masas contienen patrones de fragmentos complementarios: lo que falta en un espectro a menudo aparece en el otro. Los autores muestran que combinar tales pares espejo puede llevar la cobertura de fragmentos casi a completa, con cerca del 98% de los cortes posibles respaldados por señales experimentales reales, mucho más alto que lo observado usando una sola enzima.
Una canalización de software inteligente diseñada para datos espejo
Para aprovechar este truco bioquímico, el equipo construyó DiNovo como un flujo de trabajo de software de extremo a extremo. Primero, las proteínas de bacterias y levaduras se digieren con dos pares espejo de enzimas, y los péptidos resultantes se analizan mediante espectrometría de masas de alta resolución. DiNovo utiliza luego un módulo llamado MirrorFinder para reconocer automáticamente qué pares de espectros proceden de péptidos espejo, haciendo esto directamente a partir de los patrones de señal en lugar de basarse en conjeturas previas de secuencia. Luego, su motor principal de novo, MirrorNovo, emplea aprendizaje profundo para interpretar esos espectros emparejados, mientras que un motor de reserva basado en grafos, pNovoM2, ofrece una opción más rápida que solo usa CPU. Juntos, estas herramientas traducen picos en secuencias de aminoácidos y también examinan los espectros individuales que no formaron pares evidentes, extrayendo tanta información como sea posible.
Medir la confianza sin depender de bases de datos antiguas
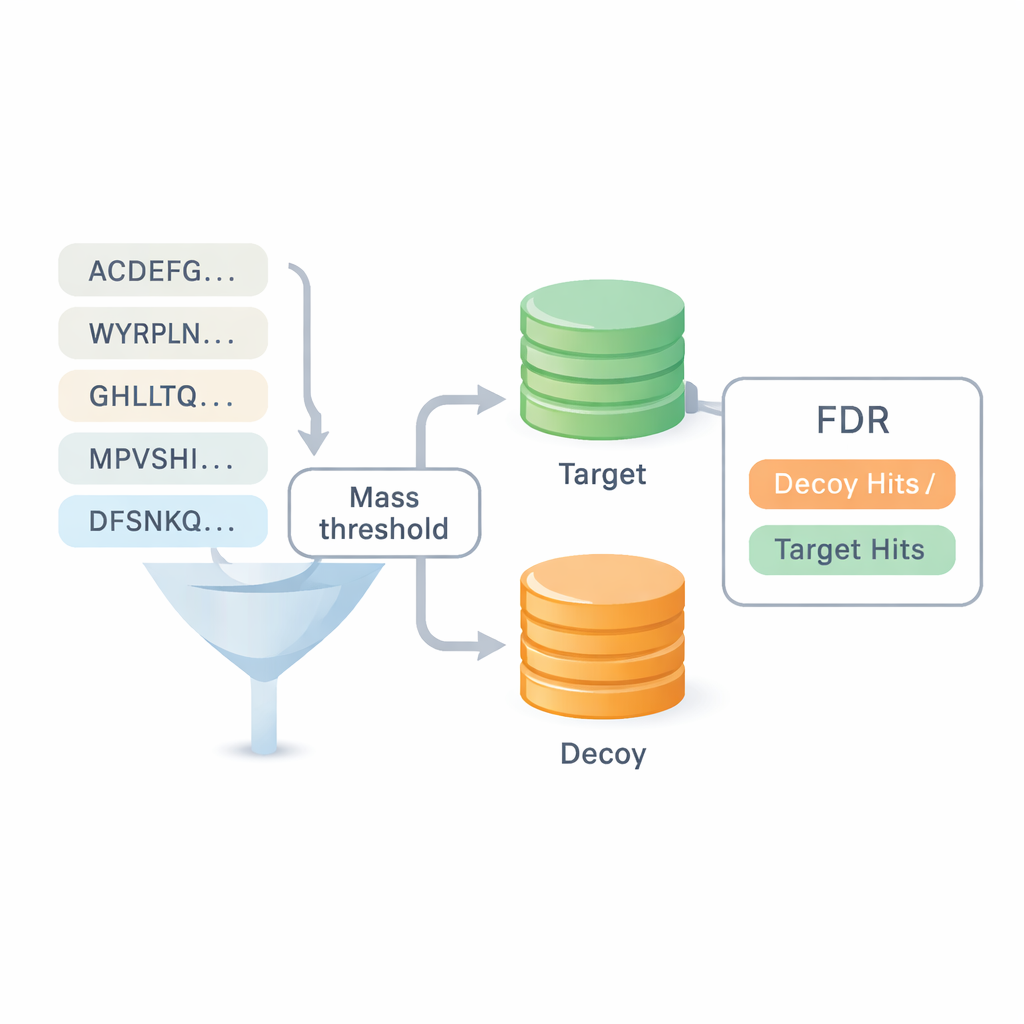
Una de las mayores preguntas en la secuenciación de novo es cuánto confiar en los resultados. La mayoría de los puntos de referencia existentes reciclan respuestas de búsquedas en bases de datos, lo que difumina la línea entre ambos enfoques y puede ocultar errores. DiNovo introduce un método de control de calidad diferente llamado mapeo objetivo-cebo. Aquí, los péptidos recién leídos se mapean a una colección combinada de secuencias proteicas reales (objetivo) y secuencias artificiales reordenadas (cebo). Al comparar con qué frecuencia los péptidos caen en el conjunto real frente al conjunto reordenado, el software puede estimar una tasa de error, o tasa de descubrimiento falso, sin apoyarse en identificaciones previas. Esto hace posible comparar DiNovo directamente con programas de búsqueda en bases de datos estándar bajo los mismos controles de error.
Lo que DiNovo entrega en la práctica
En pruebas con muestras bacterianas, de levadura y de anticuerpos, DiNovo leyó de forma constante muchos más péptidos y aminoácidos que herramientas de novo bien conocidas que usan solo una enzima. Usando dos pares espejo, produjo de 2 a 3 veces más aminoácidos de alta confianza que una configuración clásica con solo tripsina e identificó más proteínas a niveles de error similares. Al compararlo directamente con tres motores de búsqueda en bases de datos líderes, DiNovo encontró cantidades similares de aminoácidos y proteínas, y la mayoría de sus secuencias coincidieron con las de los motores de búsqueda en los mismos espectros. Los autores sostienen que este nivel de cobertura y concordancia significa que la secuenciación de novo, durante mucho tiempo tratada como un método de respaldo, puede ahora situarse al lado de la búsqueda en bases de datos como una opción seria y, en algunos casos, superior.
Panorama general: hacia una lectura proteica completa e imparcial
Para un no especialista, la conclusión es que DiNovo facilita mucho la lectura precisa de fragmentos proteicos sin estar limitado a lo que ya existe en las bases de referencia. Al duplicar o triplicar la cantidad de información de secuencia bien respaldada y proporcionar sus propios controles de error integrados, este enfoque abre la puerta al descubrimiento de proteínas poco familiares, al seguimiento de variaciones sutiles y a la exploración de mezclas complejas donde muchos componentes aún son desconocidos. En resumen, al emparejar enzimas espejo con aprendizaje profundo y estadísticas cuidadosas, DiNovo ayuda a convertir trazas espectrales ruidosas en una imagen más clara y fiable de las proteínas que sustentan la salud y la enfermedad.
Cita: Cao, Z., Peng, X., Zhang, D. et al. DiNovo enables high-coverage and high-confidence de novo peptide sequencing via mirror proteases and deep learning. Nat Commun 17, 2203 (2026). https://doi.org/10.1038/s41467-026-70224-6
Palabras clave: proteómica, secuenciación de péptidos de novo, espectrometría de masas, aprendizaje profundo, proteasas espejo