Clear Sky Science · es
Muestreo eficiente de grandes vías de transición y conformaciones intermedias en complejos proteicos sub-mesoscópicos
Observando proteínas en movimiento
Muchas de las moléculas que nos mantienen vivos se comportan menos como piezas rígidas de Lego y más como diminutas máquinas que cambian de forma constantemente. Estos movimientos impulsan procesos como la producción de energía, la reparación del ADN y la entrada de virus en las células. Experimentos como la criomicroscopía electrónica (cryo-EM) pueden ahora congelar algunas de estas conformaciones, pero no los pasos fugaces intermedios. Este artículo presenta eBDIMS2, un nuevo método informático que puede «rellenar los fotogramas perdidos» del movimiento proteico incluso para enormes máquinas moleculares que antes eran demasiado grandes y complejas para simular en un ordenador común.
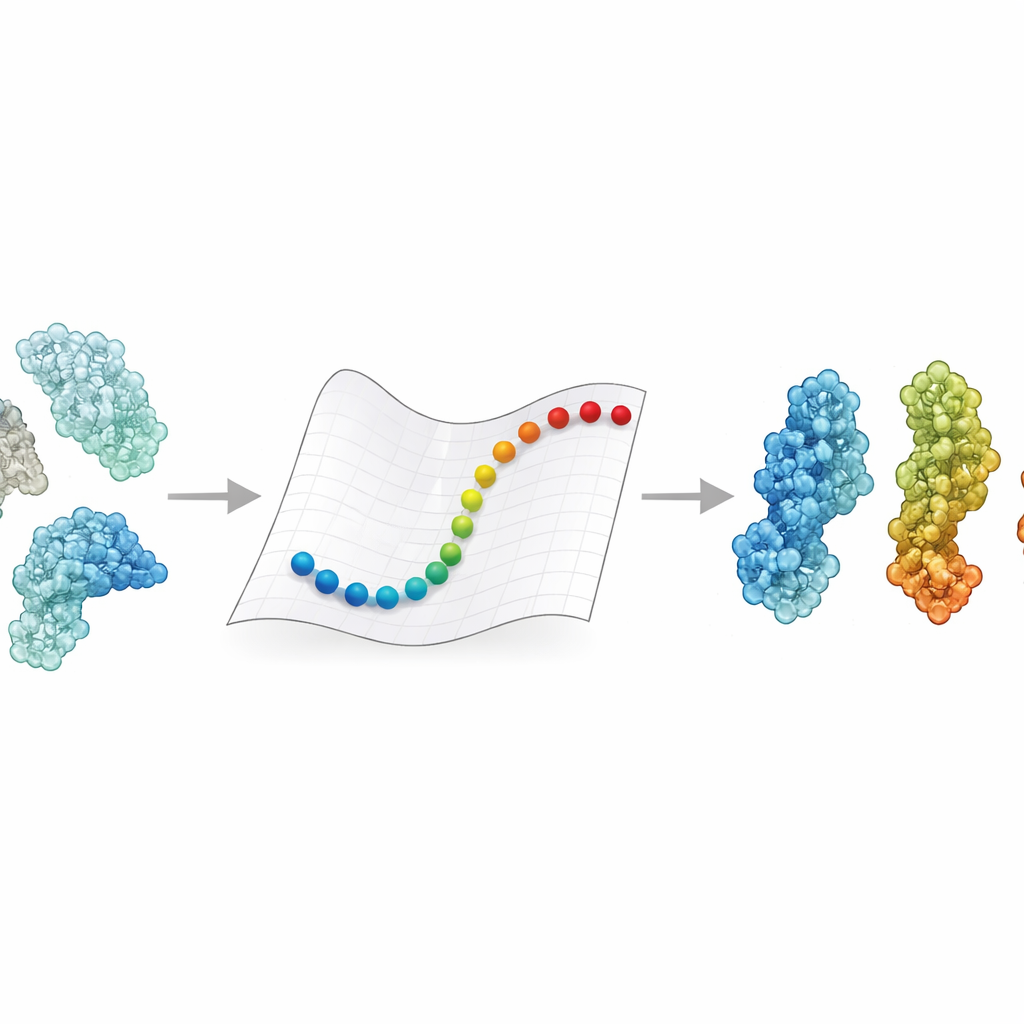
Por qué importan los cambios de forma de las proteínas
Las proteínas rara vez permanecen en una sola pose. Se abren y cierran, giran y se flexionan en respuesta a señales como cambios de voltaje, de pH o la unión de una molécula compañera. Estos desplazamientos pueden determinar si una enzima está activada o desactivada, o si un receptor atrapa un virus o lo deja escapar. Los experimentos nos dan instantáneas detalladas de algunas formas clave, y las simulaciones de dinámica molecular pueden en principio conectarlas siguiendo cada átomo en el tiempo. Pero rastrear tales movimientos para los enormes complejos que ahora revela la cryo-EM, con masas que a menudo alcanzan cientos de miles hasta millones de daltons, suele exigir superordenadores y semanas de cálculo. Como resultado, para muchos gigantes de interés médico todavía no sabemos cómo un estado se transforma en otro.
Una ruta más rápida por los paisajes proteicos
eBDIMS2 toma un atajo simplificando cómo se representa la proteína y cómo se calcula su movimiento. En lugar de seguir cada átomo, trata a cada aminoácido como un único punto conectado por resortes en una red elástica. Esos resortes capturan cómo diferentes partes de la proteína tienden a moverse juntas. El método utiliza luego dinámica Browniana —reglas matemáticas que imitan el bamboleo en un líquido— para empujar la estructura desde un estado experimental conocido hacia otro. De forma crucial, eBDIMS2 solo presta atención a las interacciones que realmente importan, usando cortes por distancia y cálculo en paralelo para reducir el coste. Esto mejora la escala del programa de aproximadamente cuadrática a casi lineal con el tamaño de la proteína. En la práctica, significa que las transiciones de ensamblajes enormes, acercándose a dos millones de daltons, pueden explorarse en horas en un equipo de sobremesa, en lugar de ser prácticamente inalcanzables.
Comprobando las rutas con proteínas reales
Para ver si estas trayectorias rápidas tienen sentido biológico, los autores reunieron conjuntos de 47 proteínas grandes y 15 complejos adicionales, sumando cientos de estructuras resueltas mayoritariamente por cryo-EM. Usaron análisis de componentes principales, una herramienta estadística que extrae las maneras dominantes en que cada proteína puede moverse, para organizar estas estructuras en paisajes conformacionales como abierto, cerrado, activo o inactivo. A eBDIMS2 se le pidió conectar pares de estados finales a través de ese paisaje. Las rutas resultantes se proyectaron de nuevo en los mismos mapas de baja dimensión, revelando si trazaban rutas suaves que pasaban cerca de intermedios observados experimentalmente. En más del 30% de los sistemas, las rutas simuladas se acercaron —a pocos angstroms— a estructuras intermedias que no se habían proporcionado como entrada. En casos exigentes como la enzima de reparación del ADN DNA-PKcs o la proteína espiga del coronavirus, las rutas a grano grueso también coincidieron bien con simulaciones a nivel atómico mucho más costosas, incluyendo dinámica molecular dirigida y ejecuciones avanzadas de muestreo mejorado.
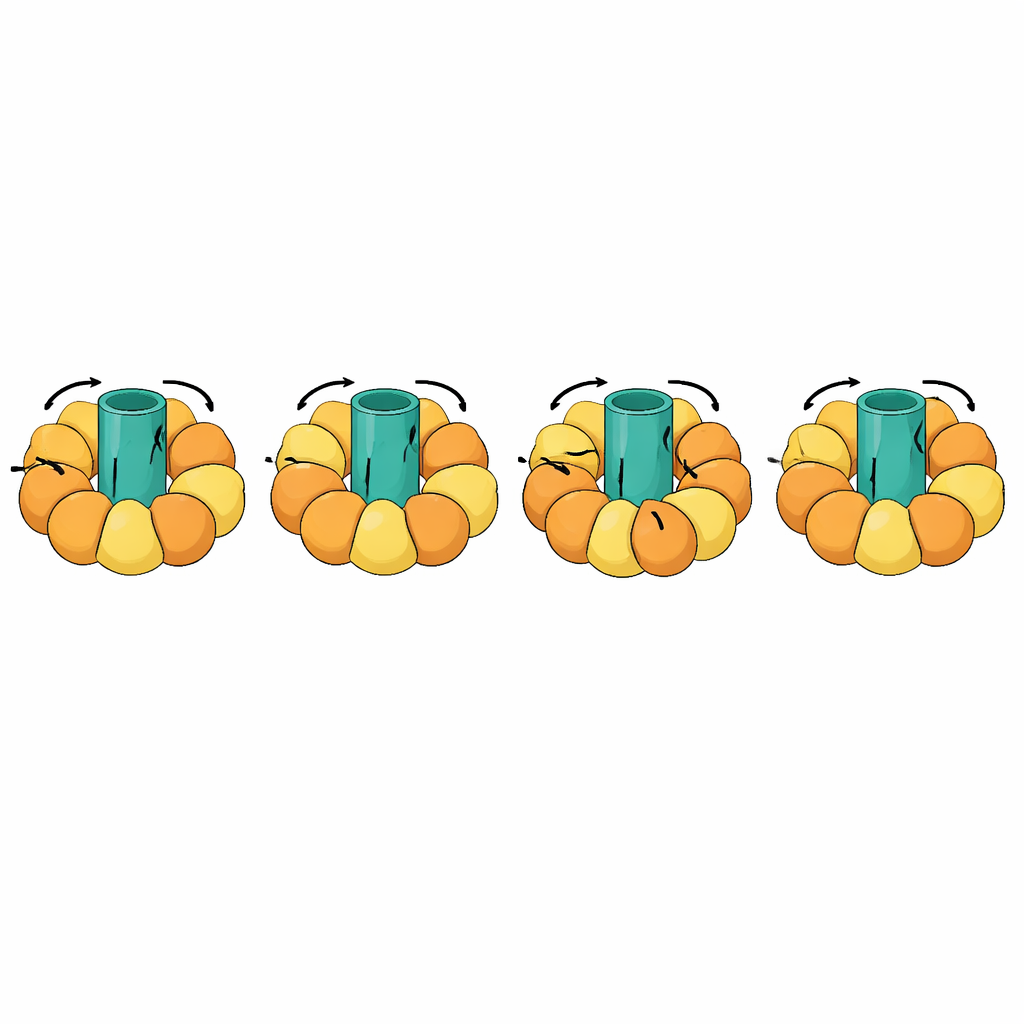
Siguiendo máquinas moleculares gigantes
Una de las pruebas más llamativas implicó máquinas rotatorias como las ATP sintasas, que generan la moneda energética celular acoplando un rotor giratorio en la membrana a movimientos de apertura y cierre en subunidades circundantes. Estas transiciones son excepcionalmente complejas: partes de la molécula deben permanecer rígidas y rotar como una unidad, mientras otras se flexionan en un ciclo coreografiado. eBDIMS2 introduce un tratamiento especial para tales piezas cuasi-rígidas y para modelos experimentales incompletos con segmentos ausentes, ambos comunes en cryo-EM. Con estas características, puede simular ciclos rotacionales completos de la ATP sintasa y otros complejos masivos como chaperonas moleculares, receptores y ensamblajes virales. En todo momento, las estructuras intermedias generadas evitan las distorsiones severas producidas por algunos métodos competidores y pueden convertirse en modelos atomísticos adecuados para cálculos de diseño de fármacos o para simulaciones más largas y detalladas.
Qué significa esto para la biología y la medicina
El estudio demuestra que eBDIMS2 puede esbozar de manera fiable las rutas principales entre formas proteicas conocidas para sistemas que estaban fuera del alcance de las simulaciones tradicionales. No sustituye las películas detalladas a nivel atómico ni proporciona energías y tiempos precisos, pero ofrece una forma rápida y físicamente fundamentada de mapear cómo podrían moverse grandes máquinas moleculares, usando solo un par de estructuras experimentales como entrada. A medida que las bases de datos estructurales se llenan con múltiples estados de grandes ensamblajes proteicos vinculados al cáncer, la infección y otras enfermedades, este enfoque ofrece a los investigadores una herramienta accesible para conectar los puntos, sugerir estados intermedios plausibles y orientar dónde buscar a continuación con métodos de mayor resolución o diseño de fármacos dirigido.
Cita: Scaramozzino, D., Lee, B.H. & Orellana, L. Efficient sampling of large-scale transition pathways and intermediate conformations in sub-mesoscopic protein complexes. Nat Commun 17, 2202 (2026). https://doi.org/10.1038/s41467-026-69809-y
Palabras clave: dinámica de proteínas, simulaciones moleculares, cryo-EM, vías conformacionales, modelado a grano grueso