Clear Sky Science · es
Activación de IRF3 en cardiomiocitos deteriora la función oxidativa mitocondrial mediante la inhibición de PGC-1α y provoca insuficiencia cardíaca
Por qué importan los corazones estresados y las células fatigadas
La insuficiencia cardíaca suele describirse como un «desgaste» del corazón, pero en su base también es la historia de una inflamación crónica y de centrales energéticas agotadas dentro de las células del músculo cardíaco. Este estudio plantea una pregunta aparentemente simple con grandes implicaciones: ¿existe un interruptor molecular único en las células cardíacas que conecte la inflamación dañina con la pérdida de producción energética —y, de ser así, puede cambiarse para alterar el curso de la insuficiencia cardíaca? Siguiendo ese hilo, los autores identifican un actor clave y muestran que potenciar de forma suave el propio programa energético del corazón puede rescatar parcialmente corazones en fallo en ratones.
Un interruptor molecular en corazones humanos enfermos
Los investigadores se centraron en una proteína llamada IRF3, conocida principalmente por ayudar a las células a responder ante infecciones virales. Examinaron tejido de personas con miocardiopatía isquémica, una forma común de insuficiencia cardíaca causada por el flujo sanguíneo reducido tras infartos. En estos corazones en fallo, IRF3 no solo estaba presente: aparecía químicamente activada en sitios específicos, señal de que impulsaba programas génicos. Al mismo tiempo, la maquinaria que permite a las mitocondrias convertir el combustible en energía mediante fosforilación oxidativa estaba visiblemente debilitada. Un patrón similar surgió en modelos de ratón de infarto: al ligarse una arteria coronaria, IRF3 en las células del músculo cardíaco se activó con fuerza y se encendieron los genes controlados por IRF3. Incluso fragmentos de ADN mitocondrial —liberados por mitocondrias dañadas y que actúan como señales internas de “peligro”— bastaron para activar IRF3 en cardiomiocitos aislados.
Apagar IRF3 protege el corazón
Para comprobar si la actividad de IRF3 en las células del músculo cardíaco realmente empeora la enfermedad, el equipo diseñó ratones en los que IRF3 podía eliminarse solo en los cardiomiocitos, dejando intactas las células inmunes y de soporte. Tras inducir un infarto, estos ratones mostraron mejor función de bombeo y menos cicatrización que los ratones normales, a pesar de tener la misma lesión inicial. En células cardíacas cultivadas en placa, silenciar IRF3 atenuó los genes inflamatorios sin alterar otras proteínas relacionadas. En conjunto, estos resultados sostienen que IRF3 dentro de la propia célula cardíaca no es un mero espectador: amplifica la inflamación y el daño estructural tras la isquemia y contribuye a la progresión hacia la insuficiencia cardíaca.
Cuando IRF3 queda «encendido», el sistema de combustible colapsa
Los autores invirtieron el experimento: crearon ratones en los que IRF3 en cardiomiocitos podía forzarse a un estado de actividad permanente mediante un ingenioso truco genético «fosfomimético». Incluso sin un desencadenante externo, estos ratones desarrollaron rápidamente una disfunción cardíaca severa, niveles altos de mensajeros inflamatorios en sangre y signos de lesión celular. Un análisis profundo del tejido cardíaco mostró que, cuando IRF3 está crónicamente activo, suprime a un coordinador maestro de la energía llamado PGC-1α. Esta molécula normalmente promueve mitocondrias saludables, la quema eficiente de grasas y el equilibrio energético celular. Con PGC-1α reducido, cayeron múltiples proteínas mitocondriales, la cadena de transporte de electrones flaqueó y las elecciones de combustible del corazón cambiaron: disminuyeron la carnitina y compuestos relacionados para la oxidación de grasas, se vio afectado el uso de cuerpos cetónicos y el manejo de la glucosa se volvió disfuncional. Incluso la relación NAD⁺/NADH —un indicador clave del balance redox celular— se inclinó en la dirección equivocada.
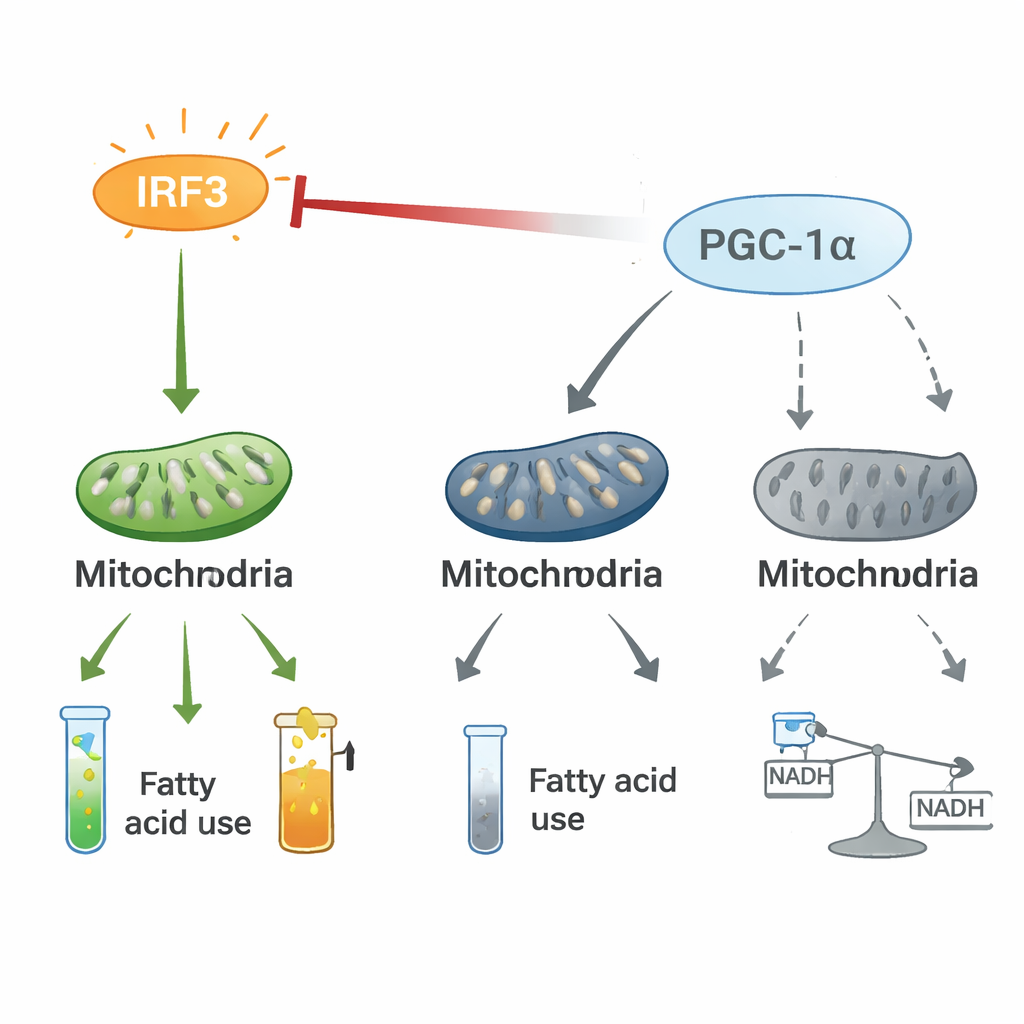
Una pugna entre inflamación y control energético
Experimentos mecanísticos revelaron que IRF3 y PGC-1α forman un eje regulatorio bidireccional. En las células cardíacas, IRF3 activado se asocia físicamente con PGC-1α y disminuye su capacidad para activar genes de la oxidación de grasas. Eliminar IRF3 eleva los niveles y la actividad de PGC-1α, mientras que aumentar PGC-1α atenúa los genes inflamatorios impulsados por IRF3 y restaura marcadores mitocondriales, incluso bajo condiciones de estrés como hipoxia o toxinas bacterianas. Trazados con isótopos estables mostraron que la activación de IRF3 redirige carbono desde la producción energética normal a través del ciclo del ácido cítrico hacia una vía alternativa, la vía de las pentosas fosfato, y perturba el flujo fluido de metabolitos. Esta pugna entre un interruptor proinflamatorio (IRF3) y un copiloto energético (PGC-1α) parece remodelar el metabolismo cardíaco de formas que favorecen la inflamación y la pérdida de energía.
Recargando suavemente las baterías del corazón
Por último, el equipo preguntó si elevar moderadamente PGC-1α podría contrarrestar el daño de IRF3. Utilizaron un vector de terapia génica dirigido al corazón para aumentar PGC-1α de forma moderada —pero no excesiva— en los mismos ratones con IRF3 hiperactivo. Este impulso modesto mejoró la función de bombeo, incrementó proteínas mitocondriales, potenció genes para la oxidación de grasas y el metabolismo de NAD, y redujo la actividad génica inflamatoria y fibrótica. En experimentos celulares, la coexpresión de PGC-1α con IRF3 activo restauró un balance NAD⁺/NADH más saludable y desplazó el uso de combustible de nuevo hacia las grasas. Para un lector general, esto significa que recargar con cuidado el “sistema de gestión de baterías” del corazón puede compensar en parte los efectos perjudiciales de un interruptor inflamatorio celular crónicamente encendido.
Qué implica esto para el futuro del cuidado de la insuficiencia cardíaca
Este trabajo sitúa a IRF3 como un nexo central entre inflamación y fallo energético dentro de las células del músculo cardíaco. En lugar de tratar la inflamación y el metabolismo como problemas separados en la insuficiencia cardíaca, el estudio sugiere que están entrelazados a través del eje IRF3–PGC-1α. Aunque estos hallazgos proceden de ratones y células, plantean la posibilidad de que futuras terapias puedan inhibir la actividad de IRF3 o fortalecer a PGC-1α y la función mitocondrial para frenar o prevenir la insuficiencia cardíaca tras un infarto. En términos simples, calmar un sistema de alarma celular sobreactivado y apoyar las fábricas de energía del corazón podría ser una estrategia combinada poderosa para mantener corazones debilitados latiendo con fuerza durante más tiempo.
Cita: Kumari, M., Evangelakos, I., Deshpande, A. et al. Activation of IRF3 in cardiomyocytes impairs mitochondrial oxidative function through PGC-1α inhibition and drives heart failure. Nat Commun 17, 2051 (2026). https://doi.org/10.1038/s41467-026-69792-4
Palabras clave: insuficiencia cardíaca, inflamación, mitocondrias, cardiomiocitos, PGC-1α