Clear Sky Science · es
TCL1A media los defectos de metilación del ADN en mola hidatiforme recurrente con variantes patógenas de NLRP7
Por qué esto importa para la salud de las mujeres
Algunos embarazos fracasan muy pronto, transformándose en una masa de tejido placentario anormal en lugar de un feto en desarrollo. Esta condición, llamada mola hidatiforme, puede repetirse en algunas mujeres y en ocasiones progresar a cáncer. Este estudio investiga una causa genética principal de estos embarazos raros pero graves y revela cómo una sola salvaguarda defectuosa en el óvulo puede desbaratar las "etiquetas" químicas que guían un desarrollo sano.
Un problema del embarazo que se origina en el óvulo
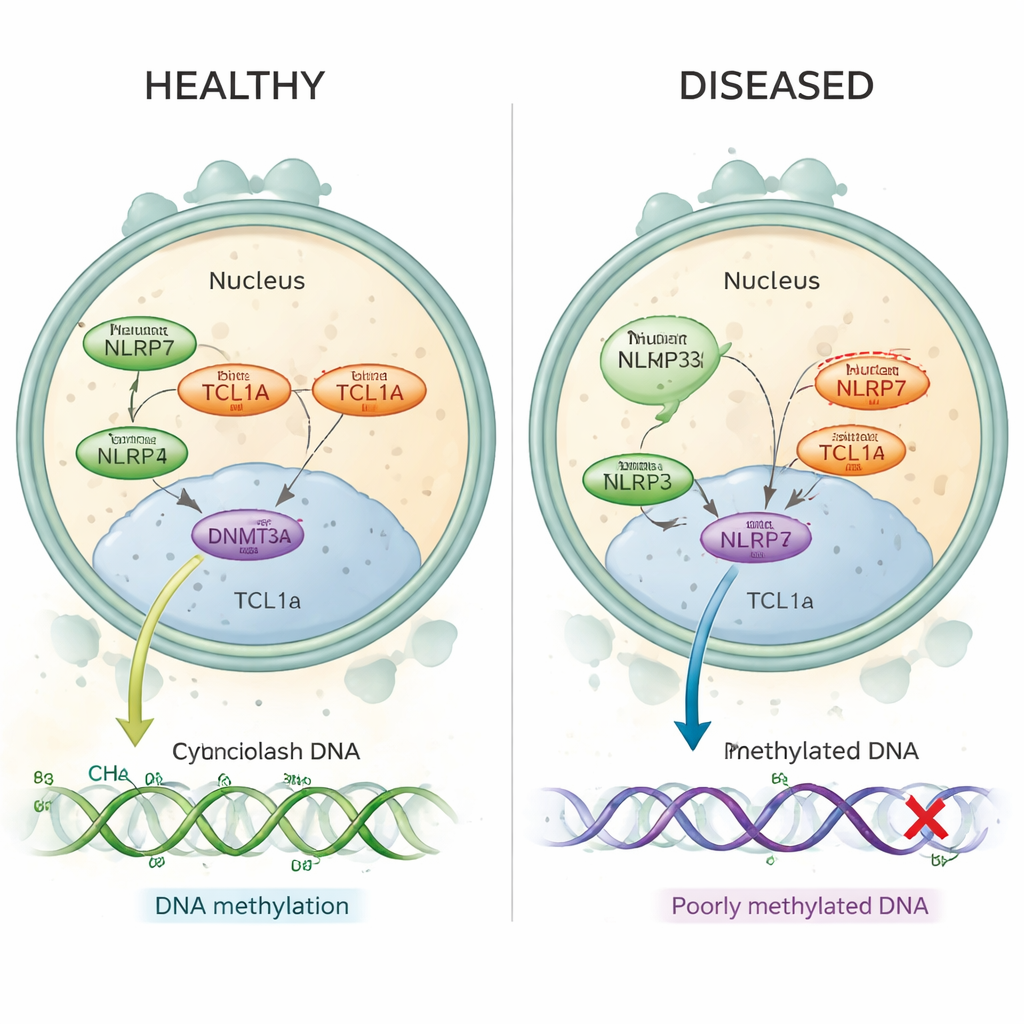
La mola hidatiforme recurrente es un trastorno en el que las mujeres sufren embarazos molares repetidos, a menudo sin embarazos normales entre ellos. Muchos de estos casos se conocen ahora por cambios dañinos en un gen llamado NLRP7, que está activo en el óvulo antes y justo después de la fertilización. En estas mujeres, tramos clave del ADN que deberían llevar "improntas" maternas carecen de las marcas normales de metilación: pequeñas etiquetas químicas que ayudan a activar o silenciar genes en el momento adecuado. Hasta ahora, los científicos no entendían cómo una proteína situada en el citoplasma del óvulo, como NLRP7, podía controlar la metilación que ocurre en el ADN empaquetado dentro del núcleo.
Encontrar una pareja que falta
Para resolver este enigma, los investigadores examinaron óvulos humanos desechados y embriones muy tempranos procedentes de clínicas de fertilidad. Aislando NLRP7 y sus socios conocidos de estas células, identificaron otras proteínas que se asocian con él. Una destacó: TCL1A, conocida en cánceres de la sangre como una proteína que puede entrar en el núcleo e interferir con las enzimas que metilan el ADN, llamadas DNMT3A y DNMT3B. TCL1A es inusualmente abundante en los óvulos humanos, lo que sugiere que desempeña un papel importante allí. Pruebas detalladas de interacción mostraron que TCL1A se une de forma fuerte y específica a NLRP7, pero no a proteínas estrechamente relacionadas, y que este complejo está integrado en una estructura mayor específica del óvulo llamada complejo subcortical materno.
Viendo el abrazo molecular
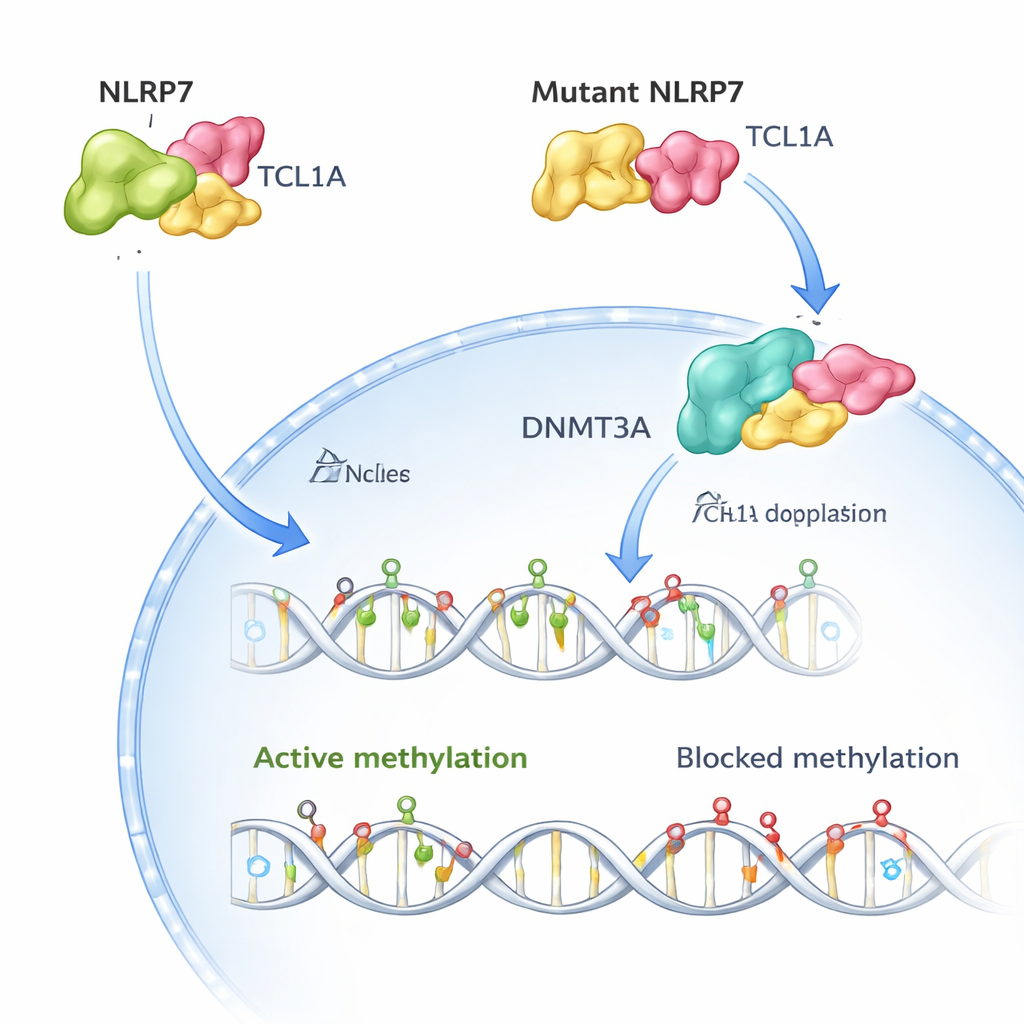
El equipo utilizó entonces criomicroscopía electrónica para visualizar la estructura tridimensional del complejo NLRP7–TCL1A. Encontraron que dos moléculas de NLRP7 se emparejan y cada una sujeta a un dímero de TCL1A a lo largo de una superficie curva de motivos repetidos. Esta disposición explica por qué muchas variantes causantes de enfermedad en NLRP7 se agrupan a lo largo de esa superficie: alterar puntos de contacto clave debilita o destruye el abrazo con TCL1A. Cuando los investigadores recrearon en células más de 50 variantes de pacientes conocidas, la mayoría de las versiones vinculadas a embarazos molares recurrentes o bien desestabilizaban NLRP7 o reducían drásticamente su capacidad de unirse a TCL1A.
Cómo las proteínas mal ubicadas alteran las marcas del ADN
En los óvulos humanos sanos, tanto NLRP7 como TCL1A se encuentran mayormente en el citoplasma, con solo una pequeña fracción de TCL1A alcanzando el núcleo. Los autores muestran que NLRP7 actúa eficazmente como un guardian: cuando puede sujetar a TCL1A, ésta se mantiene fuera del núcleo. Cuando NLRP7 está mutado y ya no puede unirse bien, TCL1A se filtra hacia el núcleo. Allí se une a DNMT3A, la enzima principal que coloca nuevas marcas de metilación en los óvulos, y atenúa su actividad. En modelos de células madre que normalmente ganan metilación a medida que maduran, la sobreexpresión de TCL1A provocó una pérdida dramática de metilación a lo largo del genoma, mientras que la coexpresión de NLRP7 rescató parcialmente este defecto. En conjunto, estos hallazgos apoyan un panorama sencillo: NLRP7 normal mantiene una "freno" de metilación (TCL1A) bloqueado en el citoplasma para que DNMT3A pueda etiquetar correctamente el ADN; NLRP7 defectuoso deja que ese freno se desplace al núcleo y bloquee el proceso.
Del mecanismo al diagnóstico
Más allá de explicar cómo surgen las molas recurrentes, el estudio sugiere una forma práctica de evaluar si un cambio recién descubierto en NLRP7 en una paciente es realmente dañino. Los autores comparan tres enfoques—pruebas de laboratorio de la unión NLRP7–TCL1A, predicciones por ordenador y herramientas genéticas estándar—y muestran que la pérdida de unión a TCL1A se alinea estrechamente con las variantes que causan la enfermedad. También descubren una variante dañina no reconocida previamente, L766R, en familias con molas recurrentes, confirmando que tanto debilita la proteína como redirige TCL1A al núcleo.
Qué significa esto en términos simples
Este trabajo revela una reacción en cadena molecular detrás de un trastorno del embarazo raro pero devastador. En esencia, los óvulos de las mujeres afectadas llevan una proteína "guardaespaldas" rota, NLRP7, que no consigue mantener a su socia TCL1A fuera del núcleo. Una vez dentro, TCL1A interfiere con la enzima que escribe etiquetas químicas vitales en el ADN. Sin esas etiquetas, la placenta temprana crece de forma anormal y no puede formarse un bebé. Al trazar paso a paso esta vía, el estudio aclara por qué ciertos cambios genéticos en las madres pueden sabotear repetidamente los embarazos y apunta hacia un asesoramiento genético y diagnóstico más precisos para las mujeres con mola hidatiforme recurrente.
Cita: Gao, Z., Liu, Q., Li, L. et al. TCL1A mediates DNA methylation defects in recurrent hydatidiform mole with NLRP7 pathogenic variants. Nat Commun 17, 2160 (2026). https://doi.org/10.1038/s41467-026-69744-y
Palabras clave: Metilación del ADN, Mola hidatiforme recurrente, NLRP7, TCL1A, Impronta genómica