Clear Sky Science · es
Ampliando el alcance de las redes neuronales de grafos con codificaciones globales
Por qué importan los enlaces a larga distancia en las moléculas
Desde nuevos fármacos hasta baterías más eficaces, muchos de los avances actuales dependen de modelos por ordenador que pueden predecir cómo miles de átomos se empujan y atraen entre sí. Una clase popular de modelos de IA, denominados redes neuronales de grafos, se ha convertido en una herramienta fundamental para esta tarea. Sin embargo, estos modelos tienen un punto ciego: en su mayoría prestan atención a los vecinos cercanos, aunque átomos distantes pueden influirse fuertemente entre sí mediante fuerzas eléctricas y cuánticas. Este artículo presenta RANGE, una forma de dotar a estas redes neuronales de una especie de visión global para que puedan “sentir” y predecir efectos a larga distancia sin volverse demasiado lentas o voraces en memoria.
Cómo la IA actual solo ve el vecindario
Las redes neuronales de grafos tratan una molécula o un material como una red de nodos (átomos) conectados por aristas (sus relaciones). En cada paso, cada nodo actualiza su estado comunicándose solo con sus vecinos cercanos dentro de una distancia fija. Repetir esto muchas veces difunde la información lentamente, pero esta estrategia tiene dos grandes inconvenientes. Primero, los mensajes pueden desdibujarse a medida que pasan por muchos intermediarios, un problema conocido como sobre-suavizado. Segundo, las vías estrechas en el grafo pueden estrangular el flujo de información, provocando sobre-compactación (oversquashing). Ambos problemas se vuelven serios al intentar captar fuerzas que actúan a muchos angstroms, como la electrostática y la dispersión en moléculas grandes o cristales. Simplemente ampliar la distancia de interacción o apilar más capas encarece los modelos y no resuelve completamente estos cuellos de botella.
Añadiendo nodos virtuales como centros de comunicación global
RANGE (Relaying Attention Nodes for Global Encoding) reorganiza este esquema al añadir un pequeño conjunto de “nodos maestros” virtuales que no corresponden a ningún átomo real. En su lugar, actúan como centros globales. Tras un paso ordinario de intercambio de mensajes entre átomos vecinos, la información de todos los átomos se recopila en estos centros mediante un mecanismo de atención: cada nodo maestro aprende en qué partes del sistema debe centrarse. Esta agregación crea resúmenes de la configuración de la molécula a una escala gruesa. En un segundo paso de difusión, esos resúmenes se reenvían a cada átomo, de nuevo usando atención para que cada átomo decida cuánto escuchar a cada nodo maestro mientras conserva su memoria local mediante auto-conexiones. Porque cada átomo se conecta directamente con todos los nodos maestros, la comunicación a larga distancia puede ocurrir en un solo paso, transformando el grafo en una red de mundo pequeño donde regiones distantes pueden influirse rápidamente y con eficiencia. 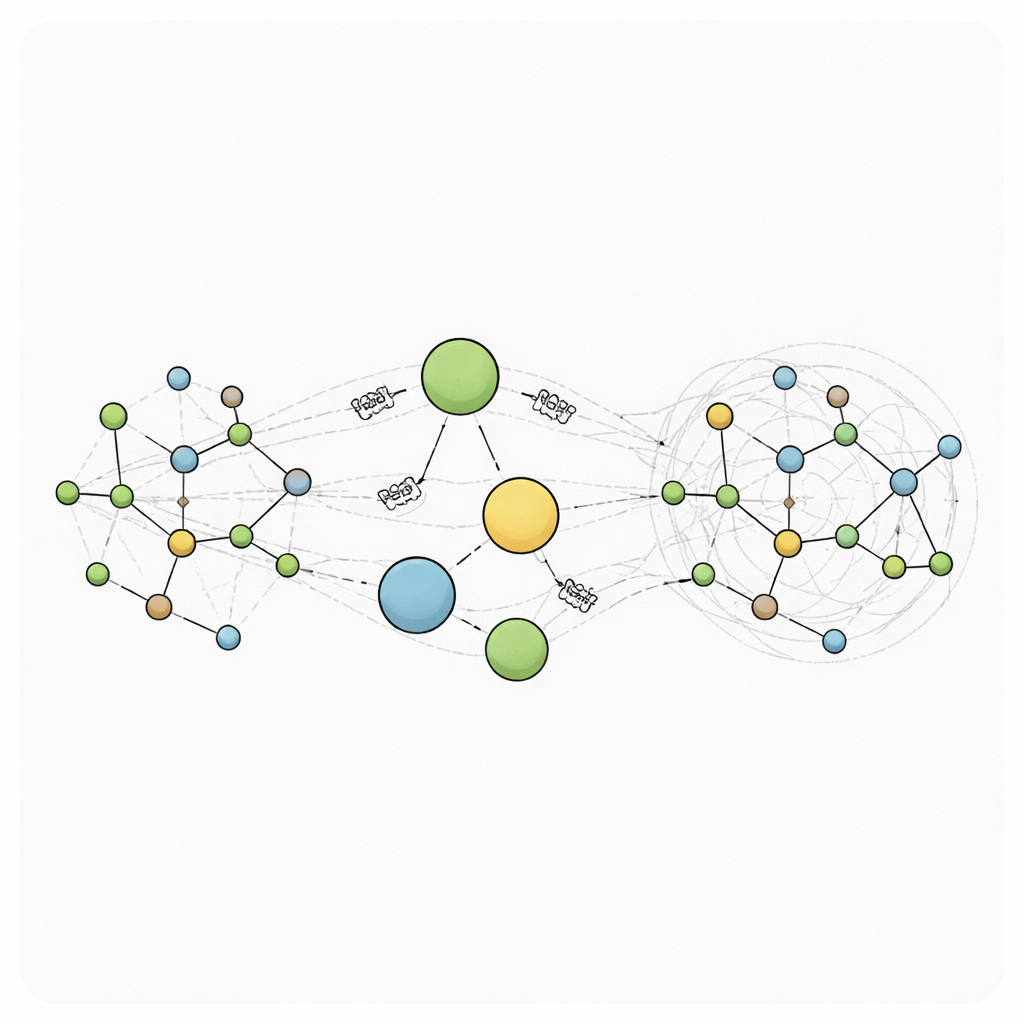
Detectando fuerzas a larga distancia que otros no ven
Los investigadores probaron RANGE acoplándolo a varios modelos de campos de fuerza moleculares de última generación y comparándolos con sus versiones originales, puramente locales. Usaron sistemas desafiantes donde se sabe que los efectos a larga distancia son cruciales: un cristal salino con un ion sodio extra que actúa como dopante, un dímero de oro aproximándose a una superficie óxida dopada, y pares de moléculas orgánicas interaccionando a distintas distancias. Los modelos estándar en gran medida no detectaron cómo las redistribuciones de carga distantes o dopantes ocultos cambiaban el paisaje energético; sus predicciones apenas variaban cuando cambiaba el entorno a larga distancia. En contraste, los modelos aumentados con RANGE capturaron correctamente las distintas curvas energéticas y pudieron extrapolar a separaciones mayores que las vistas durante el entrenamiento, con errores hasta cuatro veces menores para dímeros cargados difíciles.
Precisión sin romper el ordenador
De forma crucial, RANGE ofrece esta visión mejorada sin el elevado coste computacional de otros enfoques globales. Técnicas que toman prestado de la física, como la suma de Ewald o correcciones basadas en Fourier, requieren operaciones que crecen aproximadamente con el cuadrado del número de átomos o dependen de grandes rejillas, lo que las hace pesadas para sistemas grandes y simulaciones repetidas. RANGE, por diseño, escala linealmente con el tamaño del sistema: cada nodo maestro se conecta con todos los átomos, pero el número de nodos maestros crece de forma moderada y está controlado por un esquema de regularización que evita que se vuelvan redundantes. Los puntos de referencia en conjuntos de datos más grandes muestran que RANGE reduce de forma consistente los errores en las fuerzas predichas, incluso cuando los modelos subyacentes usan cortes de interacción cortos, y lo hace con solo un aumento moderado en tiempo de ejecución y memoria. El equipo también ejecutó simulaciones de dinámica molecular de decenas de nanosegundos en moléculas complejas, encontrando que los campos de fuerza basados en RANGE se mantuvieron estables y exploraron conformaciones y estados realistas. 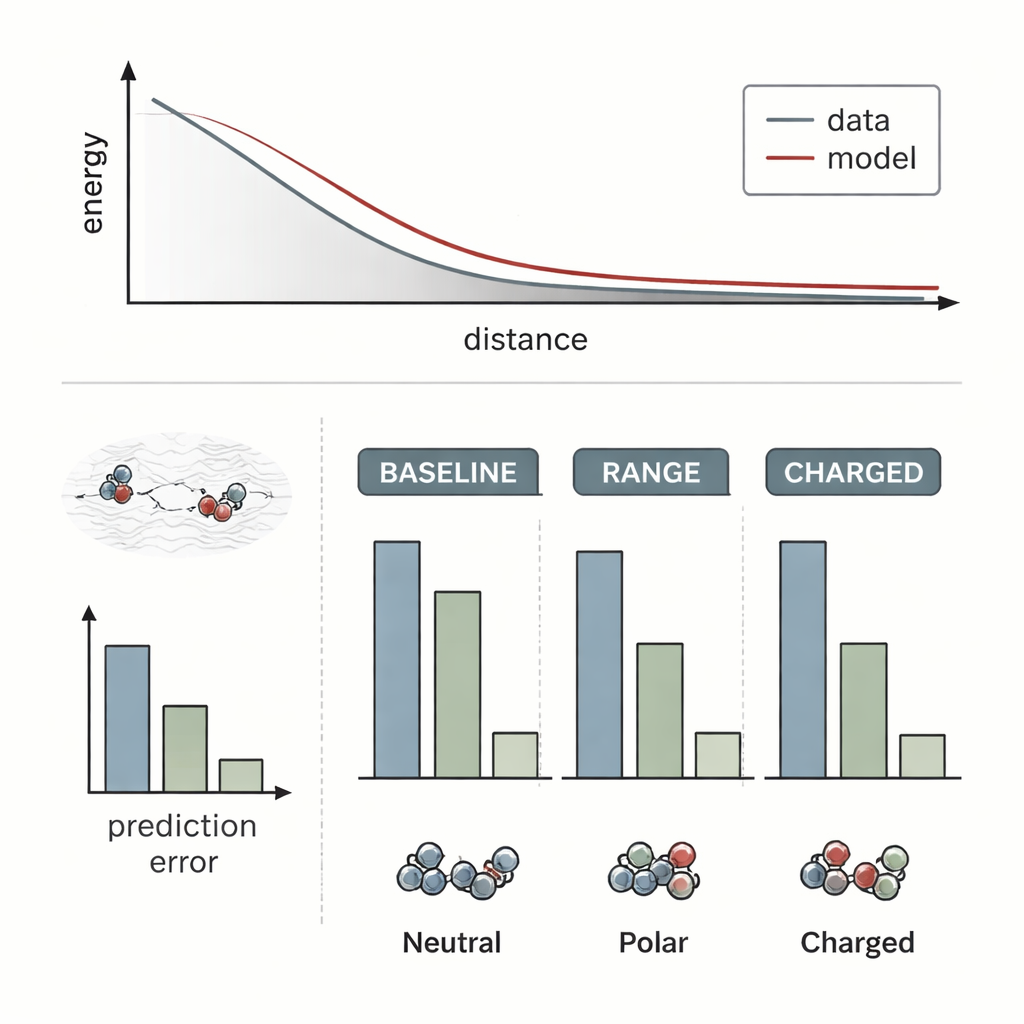
Una visión global más nítida de los mundos moleculares
Para quienes no son especialistas, el mensaje clave es que RANGE ofrece a los modelos basados en grafos una nueva forma de pensar globalmente manteniendo la operación local. Al introducir centros virtuales inteligentes y flujos de información guiados por atención, supera los cuellos de botella habituales que impiden a las redes neuronales captar efectos de muchos cuerpos y a larga distancia en moléculas y materiales. Esto se traduce en predicciones más fiables para sistemas donde regiones alejadas se influyen sutilmente entre sí —desde moléculas farmacéuticas flexibles hasta nanostructuras extendidas— sin una factura computacional prohibitiva. A medida que estos métodos se apliquen a entornos cada vez más grandes y complejos, prometen herramientas de IA que reflejen con mayor fidelidad el verdadero tejido de las interacciones físicas a larga distancia.
Cita: Caruso, A., Venturin, J., Giambagli, L. et al. Extending the range of graph neural networks with global encodings. Nat Commun 17, 1855 (2026). https://doi.org/10.1038/s41467-026-69715-3
Palabras clave: redes neuronales de grafos, interacciones a larga distancia, simulaciones moleculares, campos de fuerza aprendidos por máquina, mecanismos de atención