Clear Sky Science · es
Una referencia genérica definida por picos consensuados para el análisis de datos de ATAC-seq unicelular
Por qué importa cartografiar las «puertas abiertas» de nuestro ADN
Cada célula de tu cuerpo porta esencialmente el mismo ADN, pero las neuronas, las células sanguíneas y las tumorales se comportan de forma muy distinta. Una razón clave es que solo ciertos tramos del ADN están expuestos y "abiertos" para su uso en un momento dado. Las nuevas tecnologías unicelulares pueden ahora medir esta apertura a escala genómica, pero hasta ahora carecían de un mapa de referencia común —algo así como un atlas estándar— para comparar resultados entre experimentos y laboratorios. Este estudio construye ese mapa, llamado cPeaks, y muestra cómo puede afinar nuestra visión de los tipos celulares, el desarrollo y el cáncer.
Transformar muchos experimentos en un mapa compartido
Los autores empezaron reuniendo 624 experimentos de alta calidad que midieron la cromatina abierta —las partes accesibles del ADN— en más de 40 órganos humanos. En cada experimento, programas informáticos ya habían marcado "picos" donde el ADN estaba especialmente expuesto. En lugar de tratar cada conjunto de datos por separado, el equipo superpuso cuidadosamente todas estas listas de picos a lo largo del genoma y fusionó las regiones solapadas. Luego examinaron con qué frecuencia cada posición diminuta dentro de esas regiones fusionadas se llamó abierta en los experimentos, convirtiendo cada región en una forma característica que reflejaba con qué consistencia aparecía. Cuando una región fusionada contenía en realidad varios sitios abiertos cercanos, la dividieron en varias unidades más simples. Estas unidades —unas 1,4 millones en total— se convirtieron en los picos consensuados observados, o cPeaks, un catálogo de referencia candidato para la accesibilidad de la cromatina humana. 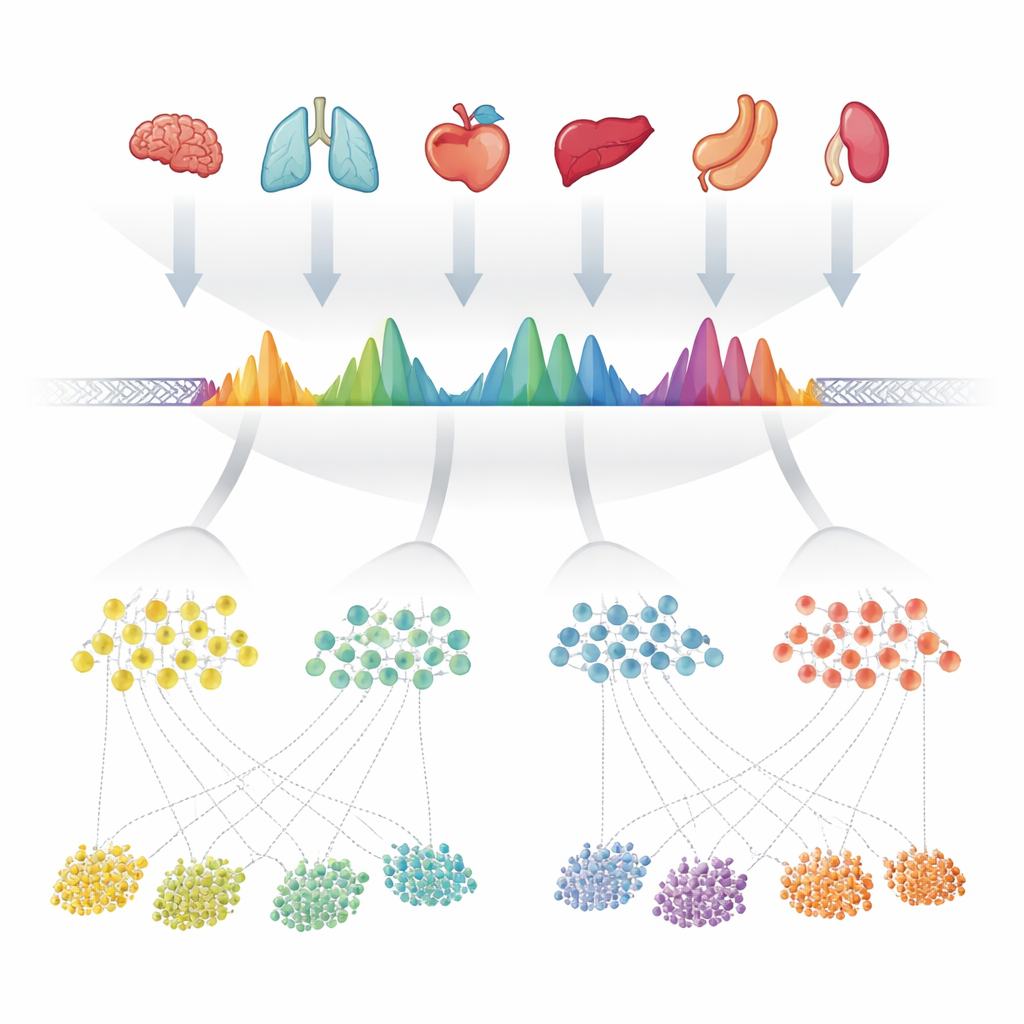
Una huella estable a través de tejidos y tecnologías
Para ser una referencia útil, estos cPeaks deben representar rasgos genuinos y repetibles del genoma, no peculiaridades de muestras o programas concretos. Los autores pusieron esto a prueba recreando sus regiones fusionadas usando solo muestras sanguíneas, solo tejidos sólidos, bases de datos públicas por separado e incluso diferentes métodos de laboratorio para sondear el ADN abierto. En cada caso, las mismas ubicaciones genómicas produjeron formas de picos notablemente similares, y la mayoría de los conjuntos de datos unicelulares que examinaron solaparon más del 90 % de sus propios picos con el catálogo de cPeaks. Las lecturas de muchos órganos se acumularon precisamente alrededor de los centros de cPeaks, mostrando que estas regiones capturan de forma fiable dónde la cromatina está abierta. En comparación con conjuntos de referencia previos basados en tecnologías relacionadas, cPeaks cubrieron más del ADN accesible detectado por experimentos ATAC-seq, y capturaron casi tanto señal como los picos definidos de novo en cada conjunto de datos —a pesar de ser fijos y reutilizables.
Enseñar a una red neuronal a encontrar regiones faltantes
Incluso cientos de muestras existentes no pueden cubrir todos los tipos celulares posibles. Para extender su mapa hacia regiones aún no observadas, el equipo recurrió al aprendizaje profundo. Entrenaron una red neuronal convolucional unidimensional sobre secuencias de ADN: ejemplos situados dentro de cPeaks observados sirvieron como positivos, mientras que regiones de fondo escogidas al azar sirvieron como negativos. El modelo aprendió a distinguir ambos con alta precisión, lo que implica que los cPeaks poseen patrones de secuencia reconocibles. Cuando los investigadores ocultaron deliberadamente picos específicos de un tejido a la vez, la red aún los recuperó solo a partir de la secuencia, incluidos sitios raros y específicos de tejido. Luego desplazaron una pequeña ventana por el resto del genoma, puntuando cada segmento y añadiendo alrededor de 280 000 nuevas regiones de alta puntuación al catálogo como cPeaks predichos, mejorando en particular la cobertura en tejidos subrepresentados en los datos originales.
Vincular regiones abiertas con genes, tipos celulares y células raras
Con una referencia más rica en mano, los autores preguntaron qué hacen estas regiones. Muchos cPeaks se sitúan cerca de sitios de inicio y término génico o se solapan con elementos reguladores conocidos como promotores, enhancers y sitios de unión para proteínas arquitecturales como CTCF. Un pequeño subconjunto es accesible en casi todos los conjuntos de datos; estos cPeaks más largos de tipo "mantenimiento" tienden a localizarse en regiones promotoras centrales de genes necesarios para el mantenimiento celular básico. El equipo también clasificó los cPeaks según lo nítidas y consistentes que son sus fronteras entre muestras, lo que refleja cuán precisamente el ADN cercano está empaquetado en nucleosomas. Las regiones con límites bien definidos están enriquecidas en familias particulares de factores de transcripción que se sabe que remodelan la cromatina y dirigen el desarrollo. Cuando se usaron los cPeaks como conjunto de características para analizar múltiples conjuntos de datos unicelulares, mejoraron la precisión del etiquetado de tipos celulares, y fueron especialmente útiles para identificar tipos celulares raros y sutiles subtipos que los conjuntos de picos previos o las mallas genómicas simples a menudo difuminaban.
Seguir el desarrollo y el cáncer usando un lenguaje común
El poder de una referencia estándar se hace evidente al comparar contextos biológicos muy distintos. Usando cPeaks, los autores reanalizaron datos unicelulares de la retina humana en desarrollo, grandes atlas de tejidos fetales y adultos, y varios cánceres. Pudieron reconstruir trayectorias de desarrollo y observar que la fracción de cPeaks con límites nítidos y "bien posicionados" tiende a aumentar durante etapas transicionales y luego disminuir cuando las células se fijan en identidades estables. Un patrón similar apareció a lo largo de las etapas tumorales: los cánceres intermedios mostraron una mayor proporción de estas regiones estructuradas, lo que insinúa un intenso remodelado regulatorio. En un tumor ovárico, los cPeaks ayudaron a revelar dos subclones cancerosos distintos con diferentes cambios en el número de copias de ADN, demostrando cómo la referencia puede exponer complejidad oculta en la enfermedad.
Qué significa esto para la investigación genómica futura
Para no especialistas, los cPeaks pueden considerarse como un conjunto estandarizado de coordenadas que marcan dónde el genoma es más probable que esté físicamente abierto y activo a través de muchos tipos celulares humanos. Alineando nuevos experimentos unicelulares de cromatina con este mapa compartido, los investigadores pueden comparar resultados entre estudios, detectar más fácilmente estados celulares raros o transicionales y comenzar a construir modelos a gran escala de la regulación génica —del mismo modo que los catálogos de genes estandarizados facilitaron el auge de los atlas unicelulares de ARN. El catálogo cPeak actual es un primer borrador que crecerá a medida que lleguen nuevos datos, pero ya proporciona un lenguaje común para describir la accesibilidad de la cromatina, acercándonos a una visión unificada de cómo el empaquetamiento del ADN guía el desarrollo, la salud y la enfermedad. 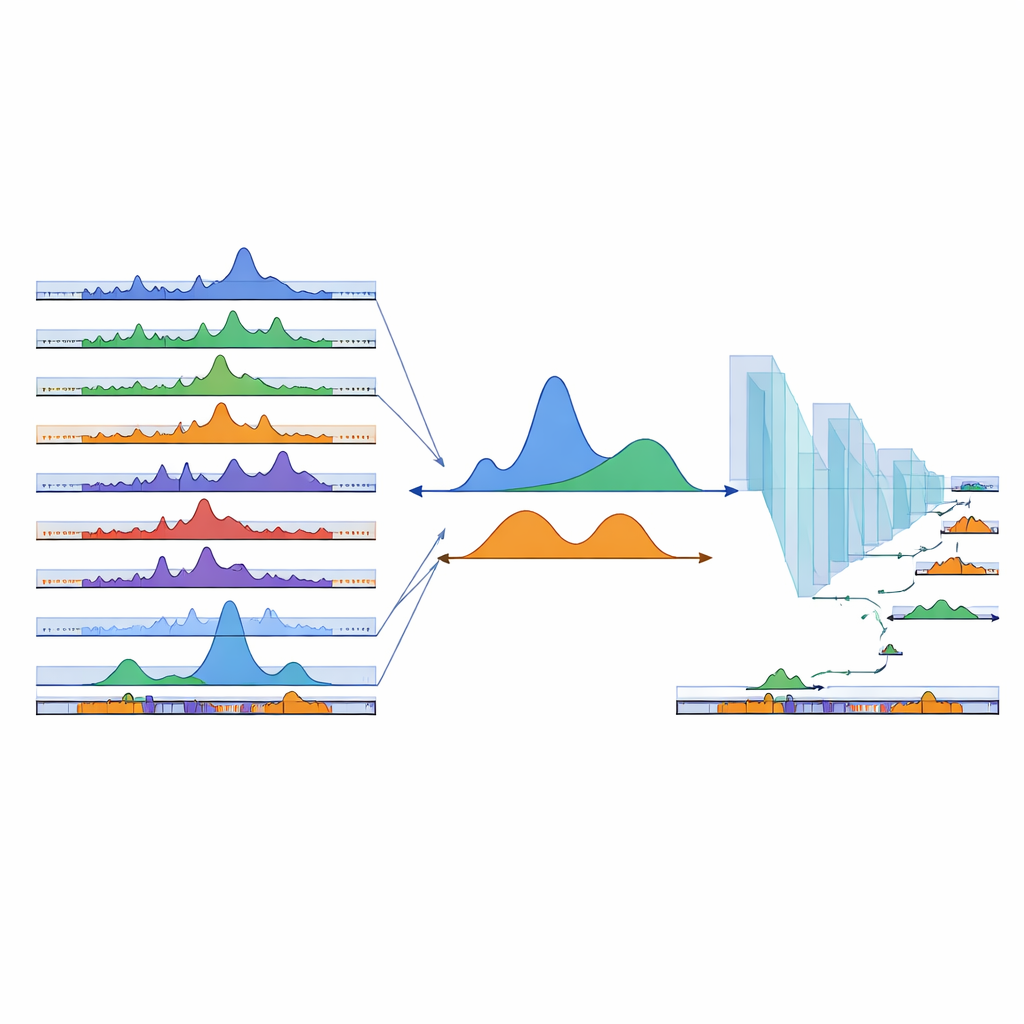
Cita: Meng, Q., Wu, X., Chen, W. et al. A generic reference defined by consensus peaks for single-cell ATAC-seq data analysis. Nat Commun 17, 2522 (2026). https://doi.org/10.1038/s41467-026-69461-6
Palabras clave: accesibilidad de la cromatina, ATAC-seq unicelular, picos consensuados, regulación génica, genómica de aprendizaje profundo