Clear Sky Science · es
Secuenciación de metilación e hidroximetilación del ADN en características cromatínicas que coexisten
Leyendo las notas químicas de nuestras células
Cada célula del cuerpo contiene el mismo ADN, pero las neuronas, las células de la piel y las células madre se comportan de forma muy distinta. Una razón es que las células escriben “notas” químicas sobre el ADN y sus proteínas de empaquetamiento, que ayudan a activar o silenciar genes. Hasta ahora, a los científicos les ha resultado difícil leer varias de estas notas juntas en el mismo tramo de ADN, lo que dejaba un vacío en nuestra comprensión de cómo actúan en conjunto. Este estudio presenta una nueva forma de leer a la vez el código genético y marcas químicas clave, revelando cómo se combinan para controlar interruptores genéticos importantes llamados enhancers.
Por qué el ADN necesita marcas de lápiz
El ADN no actúa solo. Está enrollado alrededor de proteínas llamadas histonas para formar cromatina, y tanto el ADN como las histonas pueden estar decorados con pequeños grupos químicos. Dos marcas importantes en el ADN son grupos metilo e hidroximetilo añadidos a la letra C (citosina). Estas marcas influyen en qué tan apretado está empaquetado el ADN y si los genes cercanos están activos. A grandes rasgos, las marcas de metilo suelen asociarse con el silenciamiento génico, mientras que las de hidroximetilo tienden a aparecer donde los genes están activos. Pero el efecto de estas marcas depende de su contexto local: la posición exacta en el genoma y qué marcas de histonas las rodean.
El problema de los mapas separados
Los métodos de secuenciación existentes pueden mapear marcas de metilación e hidroximetilación en todo el genoma, y otros métodos mapean marcas de histonas que señalan regiones activas o silenciosas. Sin embargo, por lo general se realizan en experimentos separados y luego se comparan por ordenador. Eso nos dice qué características suelen coincidir en la misma vecindad, pero no si realmente coexisten en el mismo fragmento de ADN dentro de una sola célula. Intentos anteriores de combinar estas mediciones dependían de tratamientos químicos agresivos que dañaban el ADN y, de forma crucial, no podían distinguir de manera fiable metilación de hidroximetilación en la misma lectura. Como resultado, los investigadores carecían de una imagen molecular nítida de cómo cooperan las combinaciones de marcas.
Un nuevo método de lectura multinivel
Los autores desarrollaron un método llamado 6-base-CUT&Tag que puede leer las cuatro bases del ADN más dos estados químicos de la citosina—normal, metilada e hidroximetilada—en fragmentos de ADN que están físicamente unidos a características cromatínicas seleccionadas. Primero, usan anticuerpos como ganchos moleculares para extraer el ADN enrollado en histonas que llevan una marca específica, por ejemplo una etiqueta de cromatina activa. Una enzima diseñada inserta adaptadores especiales, convirtiendo cada fragmento capturado en un pequeño bucle que resiste los pasos de limpieza que destruyen piezas sueltas. Un proceso químico y enzimático refinado convierte luego los distintos estados de citosina en señales de secuencia separables, que los secuenciadores modernos pueden leer. De este modo, una sola lectura indica de dónde provino el fragmento, qué marca de histona llevaba y qué citosinas estaban metiladas o hidroximetiladas.
Acercándose a los interruptores genéticos
Usando células madre embrionarias de ratón como caso de prueba, el equipo aplicó 6-base-CUT&Tag a varias marcas de histonas clave que señalan distintos tipos de ADN regulador. Se centraron en los enhancers—fragmentos de ADN que actúan como interruptores para controlar cuándo y dónde se encienden los genes. Los enhancers pueden estar en estados “activos”, “preparados” (primed) o “listos para activarse” (poised), diferenciados por marcas histónicas particulares. Los investigadores encontraron que los enhancers marcados únicamente por una etiqueta histónica llamada H3K4me1 (a menudo pensada como “preparada”) mostraban los niveles más altos tanto de metilación como de hidroximetilación del ADN, especialmente cuando se examinaban directamente en los nucleosomas unidos a H3K4me1. En contraste, los enhancers con señales adicionales de fuerte actividad o represión presentaban menos de estas marcas de ADN o mostraban un desplazamiento hacia la hidroximetilación, lo que sugiere un proceso activo de borrado de metilos.
Decodificando estados de enhancers con mayor detalle
Dado que todos los tipos de enhancer comparten la marca H3K4me1, el equipo preguntó si el patrón detallado de marcas de ADN específicamente en el ADN etiquetado con H3K4me1 podría por sí solo distinguir los distintos estados de enhancer. Entrenaron un modelo de aprendizaje automático usando los datos de 6-base-CUT&Tag para clasificar enhancers como activos, preparados o listos para activarse, basándose únicamente en cuánto metilado e hidroximetilado estaban en esa única característica histónica. Este modelo superó a otro idéntico entrenado con datos estándar de todo el genoma que no están restringidos a ninguna marca de histona. En otras palabras, leer las marcas del ADN en el contexto inmediato donde ocurren ofrece una imagen más nítida que promediar a través de todo el ADN de la célula.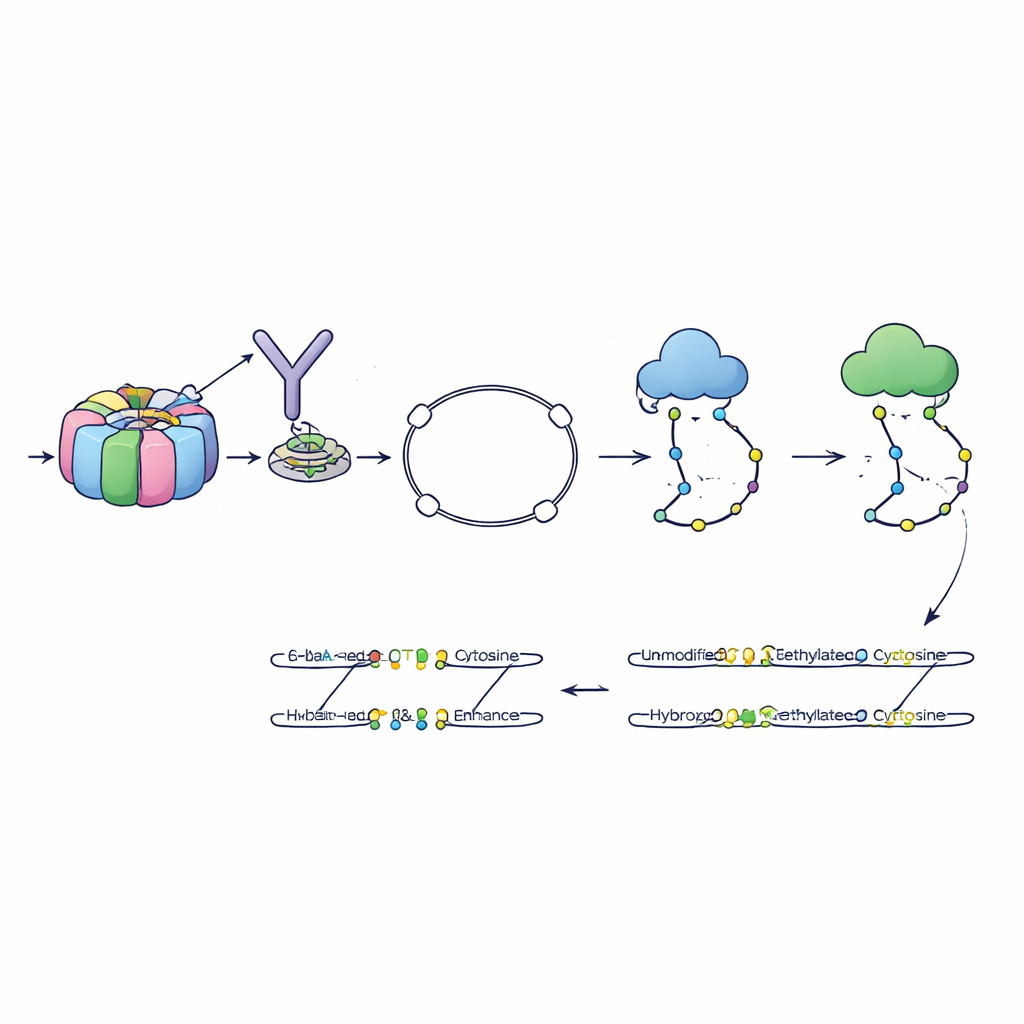
Qué significa esto para entender la identidad celular
Para un público general, el mensaje clave es que este método permite a los científicos leer varias capas de información—secuencia del ADN, marcas del ADN y marcas de histonas—en la misma molécula. Esta visión de alta resolución revela cómo combinaciones particulares de etiquetas químicas definen la disposición de los interruptores genéticos en células madre. Porque 6-base-CUT&Tag es más eficiente y menos dañino que enfoques anteriores, puede descubrir patrones sutiles que antes estaban ocultos. Con el tiempo, esta lectura multinivel de la cromatina podría ayudar a explicar cómo las células recuerdan su identidad, cómo cambian durante el desarrollo o en la enfermedad, y cómo podríamos dirigimos con mayor precisión al código regulador en terapias.
Cita: Araujo Tavares, R.d.C., Dhir, S., He, X. et al. Sequencing DNA methylation and hydroxymethylation at co-occurring chromatin features. Nat Commun 17, 2591 (2026). https://doi.org/10.1038/s41467-026-69429-6
Palabras clave: epigenética, metilación del ADN, cromatina, enhancers, células madre